中年男性,腰痛無力4年,進行性下肢無力3年
——GNE肌病
2021-06-15 08:29:18汪露露朱敏洪道俊
中國神經精神疾病雜志 2021年2期
汪露露 朱敏 洪道俊
1 資料
患者,男,31歲,裝修工人,因“腰痛無力4年,進行性下肢無力3年”于2020年8月17日就診于我院。
患者于2016年8月左右感覺腰部無力及疼痛,表現為彎腰吃力,搬重物時起身困難,疼痛在腰部活動后加重,但當時未予以重視。2017年出現左下肢無力,行走時腳尖著地,上抬費力,當時無肢體萎縮,右側下肢不受累。2018年感腰部無力較前加重,行腰椎MRI提示腰椎間盤突出,保守治療后無好轉。2019年出現右下肢無力,腳尖著地,并發現雙下肢,特別是足部肌肉萎縮。2020年感肢體無力較前加重,雙上肢受累,表現為雙上肢舉物困難,上下樓梯困難,并發現雙手肌萎縮。病程中雙小腿出現脹痛,無吞咽困難,無飲水嗆咳,無胸悶及呼吸困難,無肌肉跳動,無復視,無肢體感覺異常。自發病以來,無大小便功能障礙。
既往史和家族史:否認高血壓及糖尿病等慢性病史。自幼按計劃進行免疫接種,無特殊藥物史,無毒物接觸史。父母體健,非近親結婚,有1個哥哥和妹妹,無與患者類似疾病,無其他家族性遺傳病。
體格檢查:體溫36.2℃,脈搏87次/min,呼吸14次/min,血壓122 mmHg/87 mmHg,心律齊,叩診心界不大,各瓣膜區聽診未聞及雜音,其他內科查體未見明顯異常。神志清楚,言語清晰流利,記憶力、計算力、定向力、判斷力及理解力等高級皮層功能檢查未見明顯異常。雙側瞳孔等大等圓,直徑約2.0 mm,對光反射靈敏。雙眼瞼閉合力可,雙側眼球向各個方向運動正常,未及復視和眼球震顫。角膜反射正常,雙側面部痛溫覺無減退。雙側顳肌、咀嚼肌對稱、有力,雙側額紋、鼻唇溝對稱。懸雍垂居中,雙側軟腭上抬有力,咽反射存在。雙側轉頸、聳肩對稱有力。伸舌居中。雙手骨間肌,雙側下肢肌肉,特別是雙側脛前肌萎縮。頸屈肌力4級,雙上肢肌力5上級級,雙側髂腰肌力3級,雙股四頭肌力 5上級級,腘繩肌力 4級,左踝跖屈肌力 4上級級,左踝跖伸肌力4級,右踝跖屈肌力4級左右,右踝跖伸肌力5上級級。四肢及軀干深淺感覺正常。四肢腱反射減低。雙側指鼻試驗正常,跟膝脛試驗正常。雙側Babinski’s征和Chaddock’s征均陰性。腦膜刺激征陰性。
輔助檢查:血、尿、大便常規正常,血常規、肝腎功能、血糖、血脂、電解質、同型半胱氨酸、凝血功能、血沉、降鈣素原、風濕四項、抗核抗體、鐵蛋白、血清乳酸等均未見明顯異常。肌酸激酶:1806 U/L(正常值 43~172 U/L);肌酸激酶心肌同工酶:14.6 U/L(正常值0~18 U/L)。心電圖未見明顯異常。肝、膽、胰、脾、雙腎彩超未見異常。胸片正側位未見異常。腰椎MRI提示豎脊肌明顯脂肪化,腰椎間盤未見明顯突出(圖1A、B)。下肢MRI顯示大腿平面半腱肌和大收肌顯著萎縮伴脂肪化,半膜肌、股二頭肌長頭、股二頭肌短頭輕微脂肪化,而股四頭肌正常(圖1C、D);小腿平面右側(比目魚肌、腓骨長短肌明顯脂肪化,脛前肌輕微脂肪改變),左側(比目魚肌、腓骨長短肌、脛骨后肌、踇長屈肌明顯脂肪化,脛前肌輕微脂肪改變)(圖1E、F)。四肢神經電圖檢查示左脛神經感覺傳導未引出肯定波形,余神經運動、感覺傳導未見異常;雙側脛神經H反射未見明顯異常。肌電圖提示雙側脛骨前肌靜息時可見少量異常自發電位,小力時程正常,大力時程病理干擾相。雙側正中神經和脛神經皮膚交感未見明顯異常。
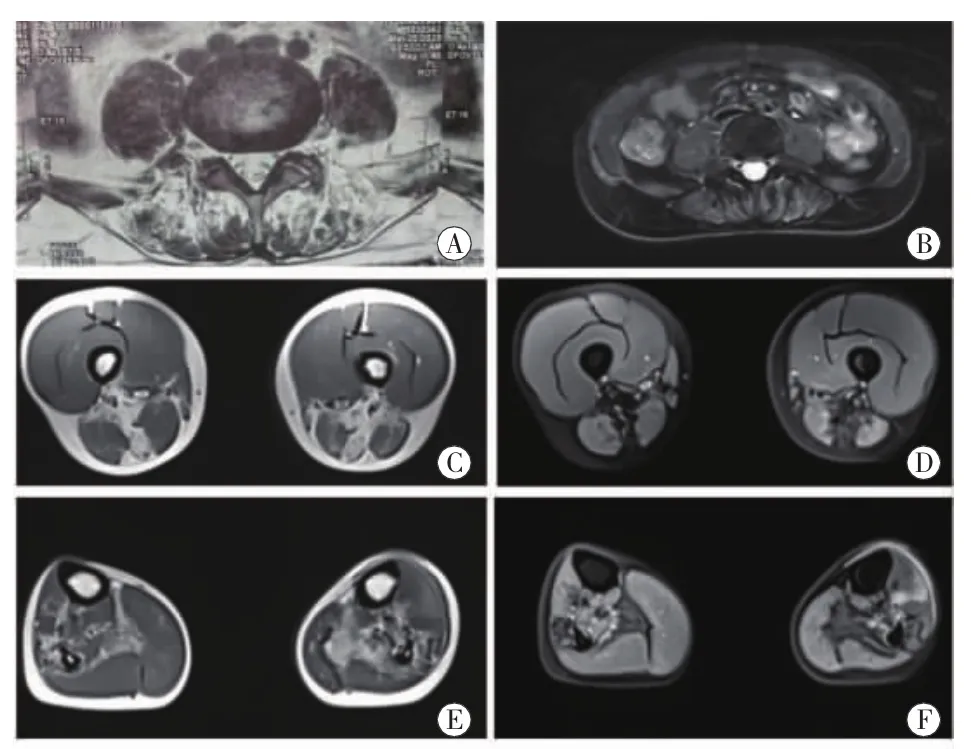
圖1 患者肌肉核磁改變 腰椎MRI顯示脊旁肌明顯脂肪化(A,T2加權),無明顯水腫(B,T2壓脂),腰椎間盤未見明顯突出。大腿肌肉MRI顯示半腱肌、股二頭肌長頭、股二頭肌短頭及大收肌明顯脂肪化,股四頭肌正常(C,T2加權),無明顯水腫改變(D,T2壓脂)。小腿肌肉MRI顯示比目魚肌、腓骨長短肌明顯脂肪化,脛前肌輕微脂肪浸潤(E,T2加權),腓骨長短肌輕微水腫改變(F,T2壓脂)。
病理檢查:經知情同意,行左脛前肌肌肉活檢。蘇木精-伊紅(HE)染色顯示肌束內結締組織中-重度增生,肌纖維直徑大小變異加大,可見成組及散在分布的小圓狀及小角狀萎縮肌纖維,部分肌纖維肥大變圓,可見部分的肌纖維出現空泡,其空泡內或周邊可見嗜堿性顆粒物質沉積,未見肌纖維壞死再生,及炎細胞浸潤(圖2A)。Gomri改良三色(MGT)染色顯示肌纖維內空泡周物質呈紫紅色,提示為鑲邊空泡(圖2B)。過碘酸雪夫(PAS)染色未見深染的肌纖維。ORO染色未見脂肪滴增多。NADH及SDH未見深染肌纖維。
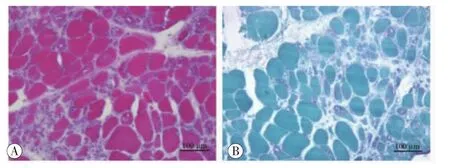
圖2 左脛前肌病理改變特點 蘇木精-伊紅染色顯示可見肌束內結締組組織中-重度增生,肌纖維直徑大小變異,可見成組及散在分布的小圓狀及小角狀萎縮肌纖維,部分肌纖維肥大變圓,可見部分肌纖維出現空泡,其空泡內可見嗜堿性物質沉積,未見炎細胞浸潤(A)。Gomri改良三色染色顯示空泡肌纖維內物質染色呈紫色,提示為鑲邊空泡(B)。
基因檢查:患者血DNA標本二代測序顯示UDP-N-乙酰葡糖胺-2-表異位酶/N-乙酰甘露糖胺激酶(UDPGlcNAc2-epimerase,GNE)基因存在復合雜合突變:分別為c.577C>T,p.R193C和 c.1196C>T,p.P399L, 兩個突變均在GNE蛋白的表位酶區,其中c.577C>T在文獻中已經報道過[1]。Sanger測序進一步證實該突變的存在,家系驗證分析顯示c.577C>T來自母親,c.1196C>T來自父親,因此符合家系共分離現象。根據臨床表現、病理改變特點、基因突變結果,該患者診斷為GNE肌病,也稱為Nonaka肌病或伴鑲邊空泡的遠端肌病。
治療:明確診斷后給予患者N-乙酰神經氨酸(唾液酸)15 g/d,溶于500 mL純凈水中,24 h內分次口服。目前已經治療4個月,但是患者肢體肌力無明顯改善,6分鐘步行試驗也無明顯變化。
2 討論
GNE肌病是一組由GNE基因突變導致的常染色體隱性遺傳性的肌病。在該疾病研究的早期發現該病患者表現突出的肢體遠端的肌無力,而肌肉纖維內出現鑲邊空泡,因此根據臨床病理特征曾用名有“遠端型肌病伴有鑲邊空泡”、“Nonaka肌病”、“鑲邊空泡性肌病而股四頭肌不受累”、“遺傳性包涵體肌病 2型(IBM2)”等[2]。然而隨著致病基因的明確,發現Nonaka肌病和IBM2肌病均為GNE基因突變所致[3-4],而且GNE基因突變導致的臨床表現也顯著地超出了既往對疾病譜的認識,故目前學界統一用GNE肌病命名[2]。GNE肌病在全世界范圍均有報告,發病率約1/100萬~9/100萬,主要是在伊朗裔猶太人和日本人群中相對常見[5]。
GNE肌病患者發病年齡跨度較大,但通常多在20~40歲,平均28.9歲[1]。典型的臨床首發表現為因脛前肌受累而導致足下垂、跨閾樣步態,逐漸累及近端、軀干肌肉導致相應的臨床癥狀,少數累及心肌和呼吸肌,一般不累及球部和面部肌肉[5]。病程進展相對緩慢,上肢發病時間平均比下肢癥狀晚4年,起病到需要輪椅的時間平均11.9年[6]。臨床上也有少數患者表現為一些不典型癥狀,例如以上肢遠端無力起病,表現為手握力下降[7];或者類似肢帶型肌營養不良發病[8],表現為肢體近端無力為主;或者類似于遠端型運動神經病或者長度依賴性運動感覺神經病,表現為周圍神經病為主[9];或者以腰部無力、背痛為首發癥狀,之后出現下肢遠端無力[10-11]。該例患者就是以腰部疼痛和無力為首發癥狀,曾被誤診為腰椎病變而延誤確診時間。
GNE肌病肌肉MRI研究顯示:在疾病早期或者不典型臨床患者中,股二頭肌短頭早期即出現顯著的脂肪化,伴隨臀小肌、脛前肌、拇伸肌、趾長肌、比目魚肌和腓腸肌內側頭的輕度受累;隨著疾病進展,股后部肌群開始受累,且股直肌、股內側肌和股中間肌也有不同程度的受累,然而股外側肌在疾病后期仍相對保留正常形態[12-13]。該例患者下肢核磁也表現出類似的受累分布模式,主要是股二頭肌短頭、大收肌、比目魚肌受累突出,但是較為特殊的是該例患者的半腱肌也在這個階段出現顯著的脂肪化,是否有一定的特異性需要進一步觀察。此外,在疾病早期的腰椎MRI已經顯示椎旁肌明顯脂肪化,與患者的癥狀相符,類似肌肉MRI在文獻中也有報告[10-11]。
GNE肌病光鏡下常表現為慢性肌病樣或肌營養不良樣的病理改變特點,具體表現為肌內衣的脂肪及纖維結締組織增生,肌纖維大小不一,肌纖維萎縮,核內移,以及伴隨個別肌纖維壞死。比較有特征意義的病理改變為許多肌纖維內出現鑲邊空泡,然而由于病程和突變位點的不同,不是每個活檢都可以看到典型的鑲邊空泡[1,5]。電鏡下在肌纖維的胞漿和細胞核內可出現直徑16~18 nm管絲狀包涵體,鑲邊空泡內含大量多層髓樣小體,顆粒細絲結構、自噬空泡及細胞碎片等,提示與自噬有關[1,5]。該例患者的肌肉活檢表現為典型的GNE肌病的肌肉病理特點,也正是典型的臨床改變和病理特點,在獲得基因診斷之前,臨床上就已經確定了GNE肌病的診斷,后續的基因結果只是驗證了我們前期的論斷。
GNE基因編碼雙功能限速酶尿苷二磷酸-N-乙酞氨基葡萄糖2-表位酶/N-乙酰甘露糖胺激酶,該酶具有表位酶和激酶兩種功能,是唾液酸生物合成中的限速酶。在GNE肌病中,GNE功能障礙導致的蛋白的唾液酸化障礙,進而介導疾病的過程[5]。目前有大于200個突變在GNE肌病中發現,其中有幾個熱點突變,Met743Thr在中東,Cys44Ser、Asp207Val、和 Val603Leu 在日本,Val727Met在印度和亞洲[5],而Asp207Val在中國最常見[1]。該例患者兩個突變位點均不是熱點突變,且有一個位點為文獻尚未報道的新突變位點。目前GNE肌病的臨床型與基因型的關系尚不明確,有研究指出典型的GNE肌病患者,突變都位于激酶區;具有當有一個突變點位于表位酶區時,患者可能出現不典型的臨床癥狀[14]。然而,另外一項研究顯示突變同時在表位酶和激酶結構域上與位于同一結構域的患者相比,臨床表現趨于更嚴重的表型[6]。該例患者的突變點位于表位酶區,因而可以解釋腰部起病的不典型臨床表現,但是否具有較嚴重的臨床表型還需進一步觀察。
由于GNE基因突變后導致唾液酸代謝通路障礙,因此臨床上很早就開始關注唾液酸產物替代療法的研究。早期在動物試驗上的研究顯示,給GNE突變小鼠補充N-乙酰神經氨酸、乙酰甘露糖胺、6'-唾液乳糖等唾液酸代謝中間產物,均能在一定程度上緩解模型小鼠的及無力癥狀和病理改變[5]。然而隨后在GNE肌病患者的臨床觀察中,上述代謝中間產物沒有對患者起到顯著的治療作用[15]。由于N-乙酰神經氨酸的半衰期非常短,所以該例患者我們采取的事24小時內不定時口服補充治療,是否有臨床效果尚需更長時間隨訪。
3 點評
肌肉影像學檢查包括肌肉超聲、肌肉CT、肌肉核磁、肌肉SPECT等檢查,這其中肌肉核磁具有多維度、視窗大、軟組織分辨率高等優勢,已經成為肌肉病變重要的輔助檢查手段。肌肉核磁通過分析肌組織的脂肪化程度、肌組織水腫改變、萎縮和肥大肌肉的分布規律、左右分布的對稱性等參數,對很多肌肉病變的定位和定性診斷具有較大的參考價值[16]。也可以用于肌肉病變的評估病情進展、治療效果、運動和康復指導等方面。甚至于對很多肌肉病具有肌肉活檢或者基因/抗體檢查的等效價值,例如杜興氏肌營養不良患者的大腿肌肉核磁出現典型的 “三葉一果”征,此種征象的特異性和敏感性都超過95%,在結合典型的臨床表型的基礎上,臨床上幾乎可以直接做出診斷[17]。因此,肌肉核磁檢查特別適合基層醫院對肌肉病的診斷,是對肌肉活檢或者分子檢查缺如時的一種重要補充。
腰痛在臨床上非常常見,引起的病因也較多,常見的原因包括腰背肌筋膜炎、腰椎間盤突出、腰肌勞損等。腰痛也常作為神經系統疾病的首發臨床表現,如CADASIL2型和CARASIL腰痛為三聯征之一[18],累及中軸的肌病如線粒體肌病、面肩肱肌營養不良、多種酰基輔酶A缺乏癥、帕金森病等。腰痛時進行腰椎部位的核磁檢查是臨床常用到的一種檢查,然而臨床醫生在閱片時關注點通常集中在脊髓及椎間盤上,甚至有時夸大腰椎病變的致病程度,試圖去解釋患者的腰痛癥狀。其實腰椎旁肌肉的變性或者無力也是導致腰痛的重要原因,本文這例患者的腰椎核磁就清楚地顯示椎旁肌肉的變性和脂肪化。因此,肌肉核磁作為一種相對新出現的檢查項目,需要引起各級醫生的重視,以提高對肌肉病變的認識和識別度。
