一個兩次多囊腎胎兒孕育史家系的臨床分析及遺傳咨詢
2019-06-03 06:19:32吳慶華蘇國玲麻希洋梅世月孔祥東史惠蓉
鄭州大學學報(醫學版) 2019年3期
吳慶華,王 參,蘇國玲,麻希洋,梅世月,孔祥東,史惠蓉
鄭州大學第一附屬醫院遺傳與產前診斷中心 鄭州 450052
多囊腎病(polycystic kidney disease,PKD)是一種臨床表型和遺傳異質性較高的疾病,分為常染色體顯性遺傳PKD(autosomal dominant PKD,ADPKD)和常染色體隱性遺傳PKD(autosomal recessive PKD,ARPKD)。最初ADPKD被認為只在成人期發病,故又被稱為成人型PKD,而絕大多數ARPKD只在嬰兒期發病,故臨床上也稱為嬰兒型PKD,目前有報道[1-2]表明ADPKD也可在兒童期甚至嬰兒期發病,而ARPKD可在出生后或更晚時期發病。ADPKD發病率估計為1/1 000~1/400[3],半數患者在70歲前進展為終末期腎衰竭,85%的ADPKD病例是由于16p13.3~13.12上的PKD1基因突變致病,15%為4q21~23上的PKD2基因突變引起。ARPKD在活產兒發病率約為1/20 000,位于6號染色體p12.3~12.2的PKHD1是其主要致病基因[4]。因患者臨床癥狀及發病時間變異大,PKD的確診需明確基因診斷,大多數患者經高通量測序技術而被診斷。本研究家系中,先證者妻子曾有兩次多囊腎胎兒孕育史。為避免先證者家庭再次孕育患兒,我們應用高通量測序技術對先證者及其妻子行遺傳性腎病相關基因篩查,對該家系相關成員行基因分析,以明確病因,為再次妊娠提供指導。
1 對象與方法
1.1研究對象先證者及其配偶妊娠兩次,均于孕中期因胎兒超聲提示多囊腎,羊水過少而終止妊娠且未留取胎兒組織標本,再次妊娠前來我院行遺傳咨詢了解生育風險。該夫婦無近親結婚史、無明確的孕期藥物或不良因素接觸史。本研究獲我院倫理委員會批準(批準號:KS-2018-KY-36),研究對象均知情同意。
1.2家系調查先證者,男,26歲,無特殊臨床表現,在我院經彩超及CT檢查提示雙側腎臟大小形態正常伴多發囊腫,尿常規提示尿蛋白1+。先證者妻子腹部彩超、尿常規及生化檢查均未見異常。先證者母親,53歲,彩超檢查提示雙腎大小形態正常伴多發囊腫,無與多囊腎相關的臨床表現及其他不適。先證者父親腹部超聲未見異常。先證者姐姐,29歲,彩超提示雙腎形態欠規整,右腎可見一大小約7 mm×5 mm的囊腫。先證者弟弟,23歲,于2 a前曾因“腹痛,腹部包塊”來我院就診,血壓高至159/101 mmHg(1 mmHg=0.133 kPa),超聲提示雙腎形態失常并多發囊腫;泌尿系計算機體層攝影尿路造影檢查發現腎形態失常,雙腎多發囊腫,左腎為著,雙腎多發結石;腎動態顯像和腎小球濾過率檢查提示腎小球濾過率下降,左右側分別為33.58及49.48 mL/min;尿常規示白細胞3+,蛋白2+;腎功能示尿素9.88 mmol/L,肌酐133 μmol/L;心臟超聲提示左室壁均勻性增厚(高血壓性心臟病樣改變)。先證者岳父母肝腎超聲未見異常。該家系遺傳圖譜見圖1。
1.3遺傳性腎病基因篩查
1.3.1 DNA提取 采集該家庭先證者及其配偶,先證者母親、姐姐及弟弟的外周血2 mL,采用北京天根公司的DNA提取試劑盒提取全基因組DNA,4 ℃保存。
1.3.2 高通量測序 采用靶向基因捕獲技術結合Illumina二代測序平臺,對先證者及其配偶行63個遺傳性腎病相關致病基因(腎臟和泌尿系發育異常、腎小球疾病、腎小管疾病、纖毛功能異常、遺傳代謝病相關的基因)的高通量測序。
1.3.3 PCR及Sanger測序 高通量測序分析發現PKD1基因為可疑致病突變位點,應用PCR擴增先證者及其母親、姐姐及弟弟全基因組DNA,并用Sanger測序進行一代驗證,應用Primer 5軟件設計引物,上游引物序列:5’-CAAGAGGCTCAAGAAACTGCCCG-3’,下游引物序列: 5’-GGCCACCAGT GAGAAGTACAGG-3’,引物由生工生物工程(上海)股份有限公司合成。采用ABIBigDye 3.1測序試劑盒對產物進行雙向測序,用ABI Sequencing Analysis 5.1.1軟件將測序結果與正常序列進行比對,驗證基因突變位點。

□:正常男性;■:患病男性;○:正常女性;●:患病女性;:先證者;:終止妊娠的患胎
圖1該家系遺傳圖譜
2 結果
2.1高通量測序結果先證者的PKHD1基因各外顯子序列可檢測到多個已知多態性變異,未檢測到已知或者疑似致病變異;其PKD1基因檢測到1個疑似致病突變:c.10678G>A(p.G3560R)雜合突變,同時檢測到多個多態性變異,未見其他明確與該疾病相關的基因突變;先證者配偶未檢測到可疑突變。
2.2PCR和Sanger測序結果對先證者及其母親、姐姐及弟弟行PKD1基因c.10678G>A突變篩查,結果確定先證者及其母親和姐姐攜帶PKD1基因c.10678G>A雜合突變;先證者弟弟未檢測到該突變,見圖2。
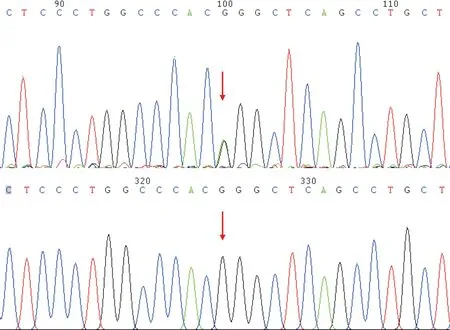
上:箭頭示先證者PKD1基因c.10678G>A(p.G3560R)雜合突變;下:先證者弟弟該位點未見突變
圖2先證者及其弟PKD1基因c.10678G>A的突變篩查結果
3 討論
絕大多數ARPKD患者在胎兒期即被診斷,常表現為雙側顯著增大的腎臟及羊水過少,由于腎臟體積過大壓迫胸腹腔而導致患兒肺臟發育異常,高達30%~50%的患兒在出生后不久即死于呼吸道功能不全,而腎功能衰竭很少是新生兒的死亡原因[5]。存活者有腎臟功能受損且通常伴腎性高血壓和門靜脈高壓,可導致新生兒期及兒童期死亡。PKHD1基因突變是導致ARPKD的主要病因,有研究表明DZIPIL可能是另一個與其發病相關的基因[6-8]。
ADPKD多在出生后人群中表現為腎臟囊腫,增大的腎臟體積與腎功能受損程度呈正相關[9-10],患者腎臟功能受損表現為尿濃縮功能下降,隨后出現腎功能不全等,還可有因感染或囊內出血導致的急性或慢性疼痛、高血壓、血尿、尿路感染、腎結石等以及腎臟外表現如肝囊腫等[11-12]。患者疾病進程變異大,絕大多數患者起病于20~40歲,僅2%~5%在兒童期確診[6],少數患者在胎兒期發病,已有相關文獻[13]報道早發型ADPKD(兒童期甚至胎兒期發病)主要由PKD1基因突變所致。ADPKD具有高度臨床異質性,即使同一家庭中攜帶相同基因突變者臨床表現也可以存在很大差別[14]。盡管ARPKD不如ADPKD常見,遺傳及臨床表型異質性不如ADPKD高,但有少數ARPKD患者可存活至兒童期甚至成人期,極少數患者可存活至老年才發病,故少數情況下ADPKD較難與ARPKD相鑒別[15-18]。
本研究家系中,先證者雙腎多發囊腫,該夫婦有兩次多囊腎胎兒孕育史且均未留取組織標本,來我院進行產前遺傳咨詢。我們首先應用高通量測序技術對先證者及其妻子行包括PKD在內的63種遺傳性腎病基因篩查,結果提示先證者PKD1基因1個疑似致病突變:c.10678G>A(p.G3560R)雜合突變,先證者妻子未檢測到該突變。先證者母親及姐姐均攜帶有PKD1基因c.10678G>A突變,而經影像學檢查明確診斷為PDK且伴有典型ADPKD臨床表現的先證者弟弟未檢測到該突變。有相關文獻[19-20]報道認為該突變可能為不致病突變或多態位點。結合本研究家系中各成員臨床表現和已有相關文獻報道,認為該突變位點致病可能性小,不是導致該家系胎兒多囊腎的原因。先證者弟弟為臨床明確診斷的成人型PKD患者,該患者拒絕行全面的遺傳性腎病高通量測序基因篩查,目前該患者病因尚不明確。
通過現有影像學及高通量測序篩查技術,大部分PKD患者都可以明確病因,但仍有少部分患者不能確定遺傳學病因[6]。本研究家系中同時有兩次多囊腎胎兒孕育史、成人型PKD患者史及多位家族成員腎臟多發囊腫表現,鑒于ADPKD和ARPKD的臨床異質性,考慮這兩類遺傳性腎病在該家系同時存在的可能性,建議該家系相關成員定期行腎臟超聲和腎功能檢查,同時先證者妻子在孕期詳細行胎兒腎臟超聲檢查以降低該家庭的生育風險。
猜你喜歡
國際太空(2023年1期)2023-02-27 09:03:42
臨床輸血與檢驗(2022年3期)2022-06-22 02:52:50
江蘇衛生保健(2021年9期)2021-03-27 16:25:04
家庭醫藥(2021年2期)2021-03-09 06:48:09
保健醫苑(2020年6期)2020-12-04 01:33:11
透析與人工器官(2020年1期)2020-11-16 01:42:34
診斷學(理論與實踐)(2020年1期)2020-04-28 07:00:24
鐵道通信信號(2019年8期)2019-10-10 05:06:00
傳染病信息(2019年2期)2019-05-17 13:16:04
中國發展觀察(2017年8期)2017-04-26 03:51:50
