1例肝豆狀核變性合并杜興型肌營養不良癥及其家系報道
2019-05-17 13:16:04閆建國陳大為徐志強王麗旻王福川
傳染病信息 2019年2期
關鍵詞:檢測
閆建國,陳大為,董 漪,徐志強,王麗旻,甘 雨,王福川, 張 敏
肝豆狀核變性是一種遺傳性銅代謝障礙疾病,由人13號染色體ATP7B基因突變造成銅沉積引起肝臟和神經系統相應癥狀,發生率約1∶10 000~1∶30 000。杜興型肌營養不良癥(Duchenne muscular dystrophy, DMD)是一種X染色體隱性遺傳疾病,主要發生于男孩,新生男嬰發生率約1∶3500。該病主要表現為骨骼肌不斷退化出現肌肉無力或萎縮,逐漸喪失行走能力,通常到20多歲因心肌、肺肌無力而死亡。患者同時并發這兩種疾病的概率極低,其出現可能與近親通婚等因素有關。本文報道1例確診為DMD合并肝豆狀核變性的罕見病例,并對其進行了家系調查。
1 病例報告
1.1 病史 先證者,男,7歲,因“發現轉氨酶高3年余”就診于中國人民解放軍總醫院第五醫學中心。4歲時體檢發現肝功能異常,間斷復查,仍異常(ALT 70~200 U/L),同時伴運動能力發育差,行走時步態異常,走路易摔倒,且狀況逐漸加重。患兒系第2胎,足月順產。
1.2 入院查體 患兒全身皮膚、鞏膜無黃染,未見瘀斑、出血點。肝掌陽性,蜘蛛痣陰性。頭顱五官無畸形,甲狀腺無腫大,心肺陰性。全腹無壓痛、反跳痛,肝脾肋下未觸及。走路步態不穩,翼狀肩胛,雙側腓腸肌肥大。肌力檢查:頸曲2級,上肢內收3級,下肢內收、伸膝2級,屈髖、背伸3級,跟腱攣縮,腱反射減低。Gowers征陽性。
1.3 實驗室檢查及其他輔助檢查 實驗室檢查:ALT 386 U/L、AST 231 U/L、CK 9288 U/L、CKMB 207.4 ng/ml、銅藍蛋白 0.02 g/L、24 h尿銅105.8 μg(參考值 15.0~ 30.0 μg)。腹部超聲:肝回聲增粗、脾大。眼科檢查:雙眼角膜K-F環陰性。心臟超聲:三尖瓣少量反流。肝臟穿刺病理檢查:考慮遺傳代謝障礙性肝病,肝豆狀核變性可能性大,Scheuer肝組織學評分G2S2-3;銅染色(+)。MRI:頭顱未見異常;雙側臀大肌及大腿肌肉脂肪浸潤伴多發水腫改變。左肱二頭肌病理診斷:骨骼肌呈肌營養不良樣病理改變。
1.4 基因檢測 全外顯子基因檢測:患兒存在2處肝豆狀核變性相關基因ATP7B純合突變(ATP7B c.2333G>T chr13:52532469 p.R778L純合突變——已知致病突變;ATP7B c.2310C>G chr13:52532492 p.L770L純合突變——疑似致病突變),家系驗證結果顯示此2處純合突變分別來自其父母(圖1)。同時發現該患兒DMD基因51號外顯子有缺失突變。線粒體全基因檢測未發現異常。全外顯子基因檢測未見明確大片段缺失及重復突變。
1.5 病例診斷 根據患兒臨床表現、查體、生化指標、輔助檢查及基因檢測,確診為DMD合并肝豆狀核變性。
2 家系調查
通過詳細問診,進行家系上溯4代調查,發現有近親結婚及多例肝豆狀核變性和/或DMD患者。此外,行父親、母親、姐姐、表兄、小姨、舅舅基因驗證,ATP7B c.2333、ATP7B c.2310、DMD基因51號外顯子的3個結果如圖1~3所示。
先證者父母為近親結婚[先證者的奶奶(Ⅱ-2)與其姥爺(Ⅱ-8)是親兄妹]。基因驗證結果顯示先證者母親(Ⅲ-3)存在ATP7B基因雙雜合突變及DMD基因51號外顯子的雜合缺失變異;先證者父親(Ⅲ-2)存在ATP7B基因雙雜合突變,無DMD基因51號外顯子的缺失或重復變異,臨床表型正常;先證者的姐姐(Ⅳ-1)與先證者存在相同位點的ATP7B基因純合突變,臨床診斷為肝豆狀核變性,同時存在DMD基因51號外顯子的雜合缺失變異,為DMD女性攜帶者。
先證者大姨(Ⅲ-4)是其母親(Ⅲ-3)的同卵雙生姐姐(外觀推測,未行基因驗證)。其兒子[先證者表哥(Ⅳ-3)]亦轉氨酶高,且伴肌酸激酶顯著升高,有DMD基因51號外顯子缺失突變,未發現ATP7B基因突變,確診為DMD。
先證者舅舅(Ⅲ-7)未發現ATP7B基因突變及DMD基因51號外顯子的缺失或重復變異;其小姨(Ⅲ-6)存在ATP7B基因雙雜合突變,未見DMD基因51號外顯子的缺失或重復變異,臨床表型正常。
先證者母系Ⅱ代親屬中有3人約50歲死于不明原因肝硬化;推測II-8、II-9均攜帶ATP7B突變;II-9攜帶DMD基因,且III-3、III-4的DMD基因異常均源自II-9;推測II-9家族有DMD和ATP7B 2個致病基因,I-4女性為兩病攜帶者或DMD攜帶+肝豆狀核變性患者。根據問診及基因驗證畫出家系圖譜(圖4)。
3 討 論
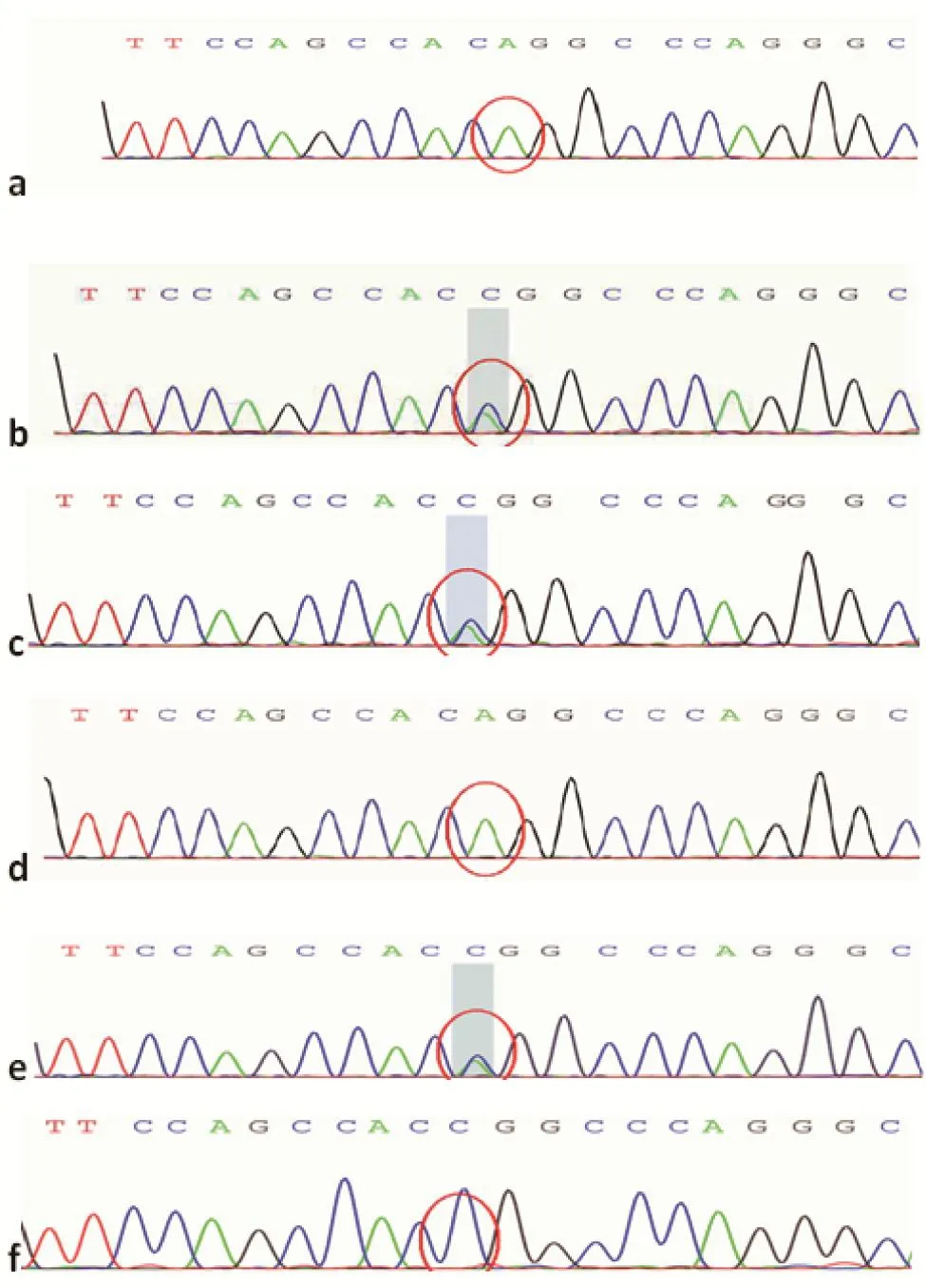
圖1 ATP7B c.2333G>T二代測序圖a. 先證者;b. 先證者母親;c. 先證者父親;d. 先證者姐姐;e. 先證者小姨;f. 先證者舅舅Figure 1 ATP7B c.2333G>T next generation sequencing
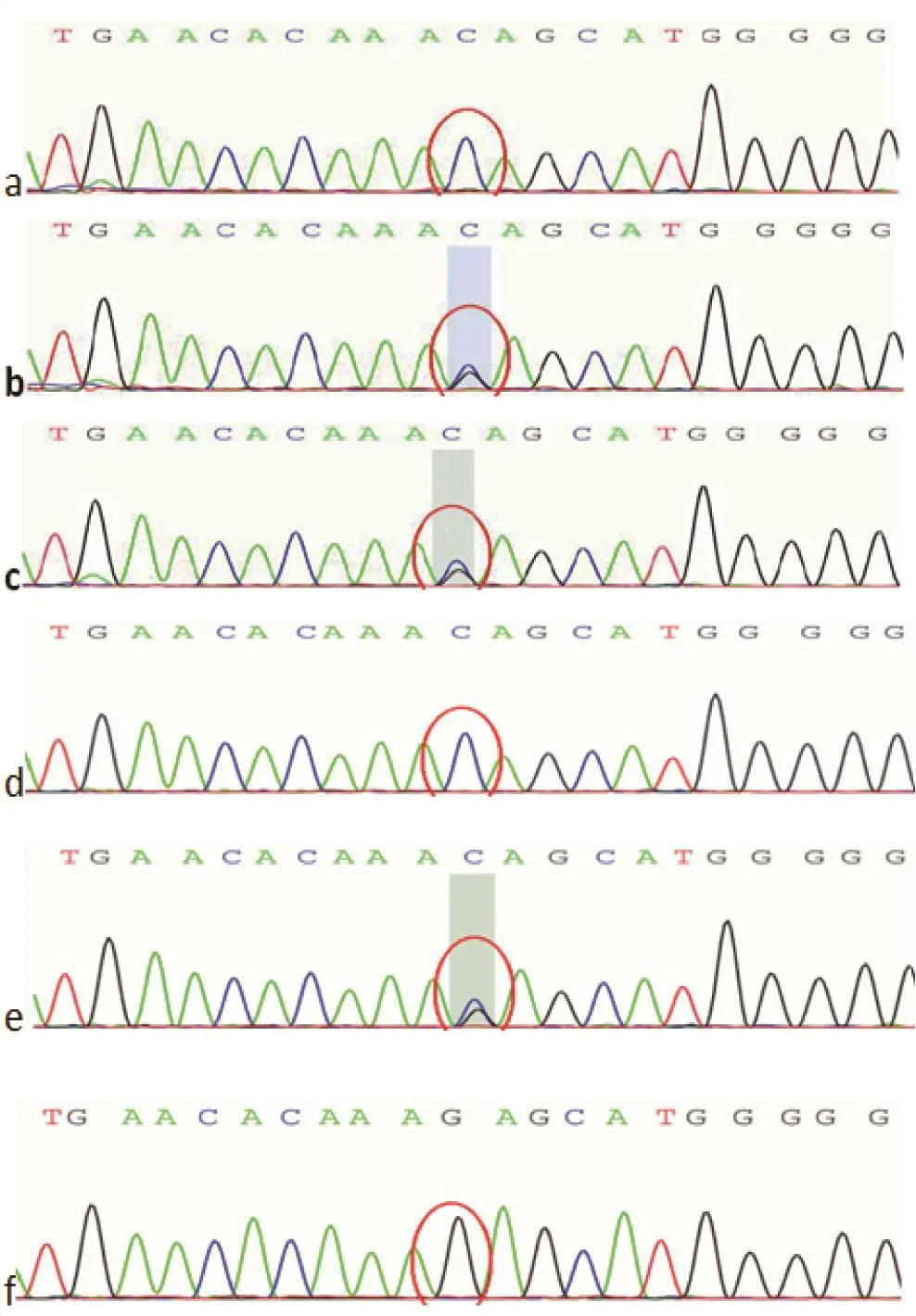
圖2 ATP7B c.2310C>G二代測序圖a. 先證者;b. 先證者母親;c. 先證者父親;d. 先證者姐姐;e. 先證者小姨;f. 先證者舅舅Figure 2 ATP7B c.2310C>G next generation sequencing
肝豆狀核變性,又稱Wilson病,是一種常染色體隱性遺傳疾病,是由編碼銅轉運所需P型ATP酶的ATP7B基因突變所致。病理學改變首先表現為肝臟中銅的毒性蓄積,銅過載后便在其他部位(例如神經系統、角膜、腎臟和心臟)中沉積,逐步進展為肝硬化、肝功能衰竭或不可逆的腦損傷。肝豆狀核變性的診斷主要通過評分系統確診(銅藍蛋白、24 h尿銅定量、肝銅、羅丹寧銅染色、K-F環、神經系統癥狀、coomb’s陰性的溶血、基因檢測)[1-2]。目前,國內研究認為Arg778Leu是中國人肝豆狀核變性的突變熱點[3]。本例先證者經基因檢測證實,存在2處純合突變,為ATP7B c.2333G>T chr13:52532469 p.R778L(已知致病突變)和c.2310C>G chr13:52532492 p.L770L(疑似致病突變),其父母、小姨、姐姐均存在相同突變位點,但因為雙雜合不發病。結合家族史及家系圖譜,可預知父系和母系Ⅰ代親屬及Ⅱ代直系親屬中均存在ATP7B基因的相同突變位點,且先證者母系Ⅱ代親屬中Ⅱ-11、Ⅱ-12、Ⅱ-13均因肝硬化約于50歲死亡,肝豆狀核變性可能性大。先證者家系中死于肝病者均已到成年,推測先證者及其姐姐肝豆狀核變性病情進展可能緩慢。
假肥大型肌營養不良癥包括DMD和貝克型肌營養不良癥(Becker muscular dystrophy, BMD),二者均是由于抗肌萎縮蛋白(dys)基因突變所致的X-連鎖隱性遺傳病。男嬰DMD的發病率約為30/10萬。DMD/BMD患者dys缺乏會引起骨骼肌細胞膜缺陷,細胞內的肌酸激酶等外漏,肌細胞壞死,脂肪組織和纖維結締組織增生[4]。DMD早期的主要表現為下肢近端和骨盆帶肌萎縮和無力、小腿腓腸肌假性肥大、鴨步和Gowers征,晚期可出現全身骨骼肌萎縮,通常在20多歲死于呼吸衰竭或心力衰竭。DMD患者病情進展快,預后差。通過典型臨床表現(雙下肢無力進行性發展、腓腸肌肥大)、肌酶顯著升高、肌電圖、肌活檢、超聲心動圖、肌肉MRI、DMD基因檢測進行確診[5]。對于典型的DMD患兒,若基因檢測已確診,則不須再行肌活檢與肌電圖。本例先證者經基因檢測證實DMD基因51號外顯子存在缺失突變;其母親、姐姐均存在相同位點的雜合缺失變異,但運動能力正常,檢測肌酶正常,未診斷為DMD,但均為DMD攜帶者;其母親及其母親的孿生姐姐為同卵雙生,其表哥(Ⅳ-3)存在DMD基因51號外顯子缺失突變,確診DMD。結合家族史及家系圖譜,可預知先證者母親及其母親的孿生姐姐(Ⅲ-4)再次生育男嬰患DMD概率高達50%。而經基因檢測,Ⅲ-6、Ⅲ-7男、女各1例均未見DMD基因51號外顯子的缺失變異,Ⅰ代親屬及Ⅱ代直系親屬男女均無DMD家族史,考慮DMD基因變異來自Ⅰ-4及Ⅱ-9女性攜帶者。可進一步完善Ⅱ代基因檢測以證實。
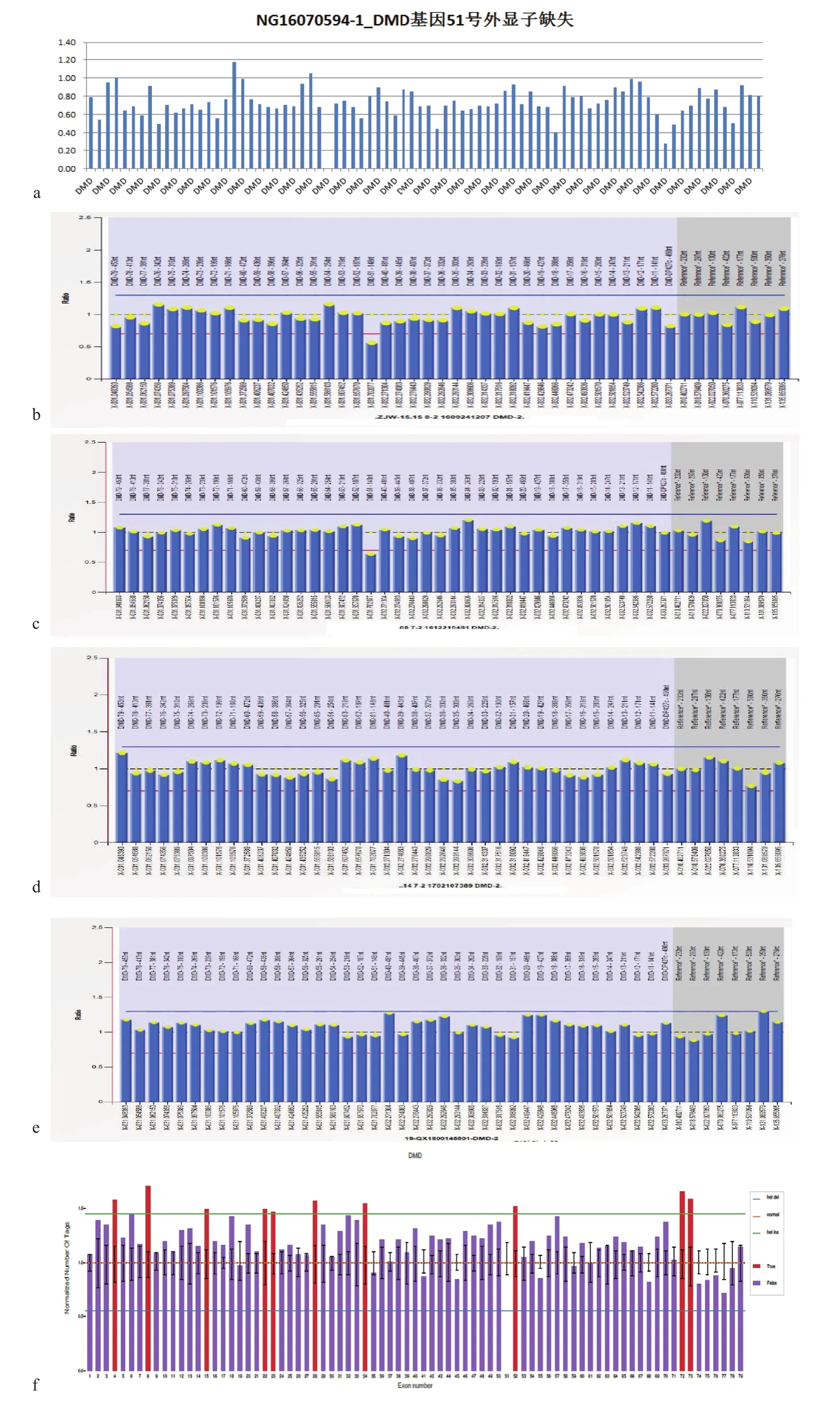
圖3 DMD基因51號外顯子測序圖a. 先證者;b. 先證者母親;c. 先證者姐姐;d. 先證者小姨;e. 先證者舅舅;f. 先證者表哥Figure 3 Sequencing of exon 51 of DMD gene
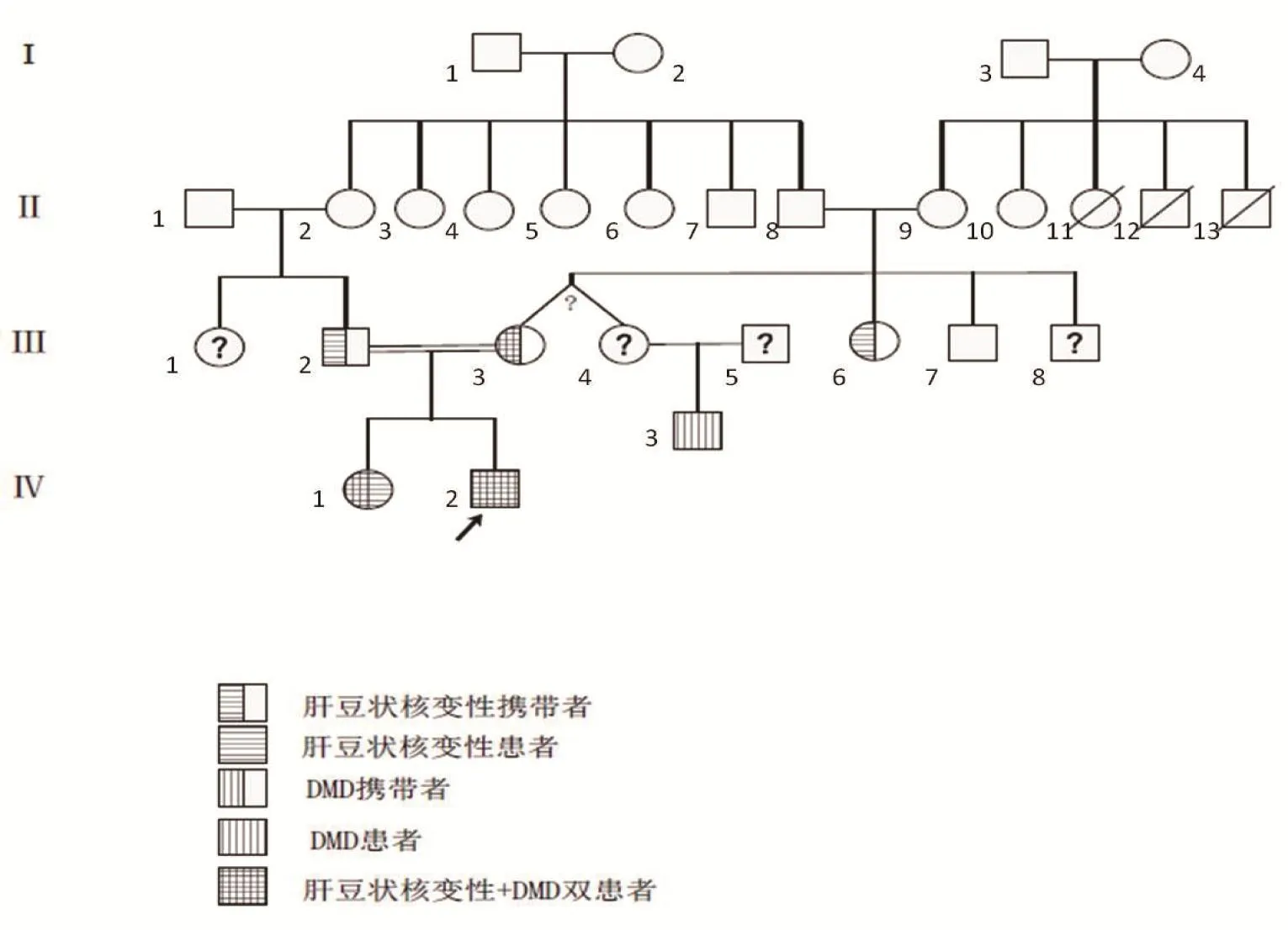
圖4 家系圖譜Figure 4 Family genetic atles
另外,通過家系分析可知,Ⅰ代父系家庭中含有ATP7B致病突變,Ⅰ代母系超家族中含有ATP7B致病突變和女性DMD基因51號外顯子缺失突變攜帶,Ⅱ代母系家族已出現多名肝豆狀核變性患者。Ⅲ代母系家族中2名女性攜帶DMD基因51號外顯子缺失突變,與Ⅲ代攜帶ATP7B致病突變的男性近親通婚,導致Ⅳ代男性出現肝豆狀核變性合并DMD。因此,Ⅳ代女性為DMD基因攜帶者,生育男嬰患DMD概率高達50%。為防止Ⅳ代女性子女的發病,強烈建議進行遺傳咨詢及產前檢查。
肝豆狀核變性與DMD均為罕見遺傳病,國內幾乎未見同時患有兩種疾病的報道。肝細胞損傷及肌細胞損傷均可導致轉氨酶的升高,轉氨酶升高的鑒別診斷對于治療意義重大。本報道通過對病史、臨床表現、體格檢查、輔助檢查及影像學、病理學、基因檢測的總結分析,最終才獲得確診。由于DMD尚無有效治療方法,故應以預防為主。而高效、準確的基因檢測手段有助于進行遺傳咨詢及產前診斷,避免患病兒出生,為預防本病提供關鍵的技術支持[6]。隨著基因編輯技術的快速進展,在基因診斷的基礎上進行有效的基因治療成為可能,為先天遺傳性疾病的治療帶來希望[7-9]。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
