脂質沉積性肌病合并橫紋肌溶解的臨床與病理特征(附1例報告)
2011-06-02 03:11:02賴鴻陳恬丁衛江
中國神經精神疾病雜志 2011年7期
賴鴻 陳恬 丁衛江
脂質沉積性肌病(lipid storage myopathies,LSM)是肌細胞內脂肪酸代謝障礙使脂質蓄積所致的一組代謝性肌病,其主要臨床表現為肌無力和運動不耐受[1]。橫紋肌溶解(rhabdomyolysis,RM)是指由于肌纖維細胞膜的破壞,肌纖維溶解,肌細胞的成分進入血液和尿液[2]。近年來國內確診的LSM病例數逐年增加[3],但合并RM者目前尚未見報道,我院最近確診1例,現對其臨床及病理學資料分析如下。
1 資料

圖1 大量篩狀空泡纖維,并見大量肌纖維壞死(HE染色200×)
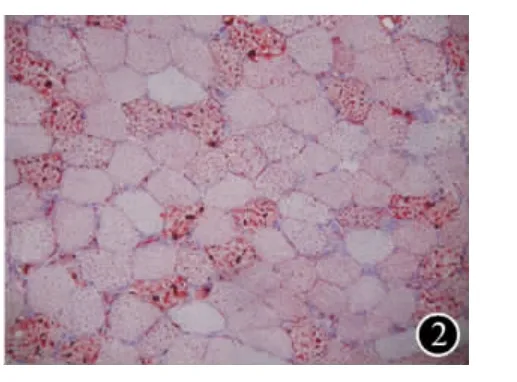
圖2 大量肌纖維內脂滴顯著增加(ORO染色200×)
患者男,30歲,漢族。約兩年前開始無明顯誘因出現運動不能耐受,做一般家務持續30 min左右即感疲勞,曾診斷“抑郁癥”給予抗抑郁藥(具體不詳)治療2周,無明顯療效。患者本次因“上腹痛脹、反復嘔吐咖啡色液體半天”于2010年3月8日入南昌市第二醫院普外科住院,診斷“上消化道出血”,經抑制胃酸分泌及對癥治療1~2 d后癥狀緩解,但1周后無明顯誘因出現雙下肢肌痛和無力,進行性加重,蹲起、上樓及行走困難,并漸波及雙上肢及頸部,雙上肢平舉費力,不能持物,抬頭困難,咀嚼及吞咽困難,并出現一過性棕色尿。患者既往有“十二指腸球部潰瘍”,多次因上消化道出血住院治療。家族中無類似疾病史。體格檢查:體溫:36.6℃,脈搏:78次/分,呼吸:20次/分,血壓:110/70 mm Hg(1 mm Hg=0.133 KPa)。神志清晰,兩眼球運動自如,雙側眼輪匝肌、咬肌、頸肌肌力Ⅳ級,咳嗽聲弱,進食有時嗆咳,懸雍垂居中,兩側軟腭運動可,四肢近端肌力Ⅱ級,遠端Ⅳ級;四肢肌肉輕-中度肌萎縮,肌張力下降,腱反射減弱,病理反射未引出,深淺感覺正常,腦膜刺激征(-)。實驗室檢查:血常規:白細胞14.8×109/L(正常值4~10×109/L),中性粒細胞0.765(0.50~0.70),淋巴細胞0.117(0.20~0.40),其余在正常范圍。肝功能、腎功能均大致正常。血電解質:K+3.11mmol/L(正常值3.5~5.5 mmol/L)、Na+140.25 mmol/L(136 ~ 145 mmol/L)、Cl-93.37mmol/L(95~108mmol/L)、Ca2+2.5 mmol/L(2.1~2.9 mmol/L)。3月17日肌酶譜:CK 14927 U/L(正常值24~192 U/L),GOT 1958 U/L(0~40 U/L),LDH 4002 U/L(15~220 U/L),α-HBDH 9567 U/L(72 ~182 U/L),CK-MB 225 U/L(0 ~25 U/L)。24 h尿量1500 mL(正常值1000~2000 mL),尿常規正常。心電圖:竇性心動過速。3月22日肌電圖示:肌源性損害。3月23日行右肱二頭肌活檢,HE染色示肌纖維大小基本一致,可見大量篩狀空泡肌纖維及壞死肌纖維,未見肌分裂、核內移及肌核增多,肌內衣輕度增加,未見灶性炎性細胞浸潤,小血管結構未見異常(圖1);ORO染色肌纖維內見大量脂滴(圖2);改良Gomri(MGT)染色未見典型不整紅邊纖維;過碘酸-希夫(PAS)染色見空泡纖維糖原輕度增多;還原型輔酶Ⅰ四氮還原酶(NADH-TR)染色示空泡纖維以Ⅰ型為主。根據患者的臨床表現、CK水平、肌電圖和肌肉病理檢查結果,確診為LSM合并RM。給予糖皮質激素及左旋肉堿治療,輔以維生素B2、輔酶Q10、三磷酸腺苷二鈉片口服。治療1周后患者肌痛及肌無力癥狀迅速改善。2周后復查CK 79 U/L,GOT 35 U/L,LDH 602 U/L,α-HBDH 628 U/L,CKMB 22 U/L。隨訪至第3個月,患者四肢近端肌力恢復至Ⅳ-級,遠端肌力Ⅴ-級;隨訪至第6個月,患者基本恢復,四肢近端肌力Ⅴ-級,遠端肌力Ⅴ級。
2 討論
LSM是一組脂肪代謝性肌病的總稱。自從1973年首例被描述以來,現在至少有14種不同的有關線粒體游離脂肪酸代謝的基因缺陷被認識,其中大部分以骨骼肌受累為主[4]。LSM主要臨床類型包括①肉堿缺陷病:分為原發性和繼發性;②肉堿轉運缺陷:分為肉堿棕櫚酰轉移酶Ⅰ(CPT-Ⅰ)缺陷病、CPT-Ⅱ缺陷病和肉堿-脂酰肉堿轉位酶缺陷病;③脫氫酶缺陷:包括不同長度脂肪酸β-氧化障礙;④功能酶缺陷:包括長鏈脂酰3-羥脂酸-CoA脫氫酶缺陷病和β-氧化通路的線粒體三功能酶缺陷病;⑤其他:包括三脂酰甘油累積病伴長鏈脂肪酸氧化障礙和電子傳遞黃素蛋白、輔酶Q10缺陷病等[5]。國內目前已有大量LSM病例報道,主要臨床表現為肌無力、運動不耐受,CK多數輕-中度升高,肌電圖正常或呈肌源性損害,對糖皮質激素和左旋肉堿治療敏感。LSM易誤診為多發性肌炎,也有誤診為心肌炎、肝炎者,本例誤診為抑郁癥屬少見。由于無分子生物學證據,國內報道的LSM具體病因一直不明,一般認為是肉堿缺乏。近期有文獻報道中國人核黃素反應性LSM多是由ETFDH基因突變所致[6]。本例患者治療同時合用了糖皮質激素、左旋肉堿及維生素B2等,并且未行基因檢測,所以具體病因也不能明確。
RM的病因分創傷性和非創傷性兩大類。非創傷性病因包括劇烈運動、肌肉缺血缺氧、感染、體溫變化、代謝和電解質紊亂、藥物和毒物,以及肌肉遺傳性代謝缺陷病等[2]。RM的發生機制包括肌細胞膜的直接損傷(如外傷)和肌漿內ATP耗竭導致肌細胞內鈣離子濃度升高兩種[7-8]。肌細胞內鈣離子由一系列的通道和泵等嚴格調控,當肌肉處休息狀態時維持低水平的鈣濃度,當肌動蛋白粘合和肌肉收縮時增加鈣濃度。ATP耗竭會損害這些泵的功能,使肌細胞鈣離子濃度持續升高,導致肌肉持續收縮和能量耗竭以及激活鈣依賴性蛋白酶類和磷脂酶,結果導致肌原纖維、細胞骨架和膜蛋白被破壞,細胞內物質被溶酶體溶解,最終肌細胞崩解[9]。
肌無力、肌痛、腫脹、棕色尿及大量肌纖維壞死而無血尿是RM的特征[2]。反復的RM通常提示肌肉本身存在潛在性代謝缺陷,包括LSM、糖原累積病和線粒體病等[2,5]。肌病患者在劇烈運動、麻醉、服用肌毒性藥物及病毒感染等時易出現RM[2]。本例患者的突出臨床特點表現為在平常運動不耐受的基礎上,短時間內出現明顯肌無力,伴棕色尿及CK顯著升高,其出現RM的原因可能與上消化道出血后患者禁食及合并低鉀血癥等有關。
當懷疑RM時,尤其是反復出現肌紅蛋白尿,肌活檢及免疫組化染色可以提供特異性診斷。肌活檢時間一般主張在發生臨床癥狀數周或數月后,因為早期肌肉活檢標本可能是正常或僅有肌纖維壞死[9]。本例患者肌活檢時間在臨床癥狀出現的第8天,但仍能顯示特異性改變,在肌纖維內脂滴顯著增加的基礎上出現大量壞死肌纖維是其肌肉病理特征。
[1]Bruno C,DiMauro S.Lipid storage myopathies[J].Curr Opin Neurol,2008,21(5):601 - 606.
[2]Xavier Bosch,Esteban Poch,Josep M.Grau.Rhabdomyolysis and acute kidney injury[J].N Engl J Med ,2009,361(1):62 - 72.
[3]沈宏銳,胡靜,趙哲,等.脂質沉積性肌病的臨床及病理學特點[J].臨床神經病學雜志,2009,22(3):178 -180.
[4]Engel AG,Franzini-Armstrong C.Myology[M].3rd ed.New York:McGraw-Hill,2004:1587 - 1621.
[5]沈定國.肌肉疾病[M].第14卷.北京:人民軍醫出版社,2007:373-376.
[6]Wen B,Dai T,Li W,et al.Riboflavin-responsive lipid-storage myopathy caused by ETFDH gene mutations[J].Neurol Neurosurg Psychiatry,2010,81(2):231 -236.
[7]Giannoglon GD,Chatzizisis YS,Misirli G.The syndrome of rhabdomyolysis:pathophysiology and diagnosis[J].Eur J Intern Med ,2007,18(2):90 -100.
[8]Wrogemann K,Pena SD.Mitochon-drial calcium overload:a general mechanism for cell-necrosis in muscle diseases[J].Lancet,1976,1(7961):672-674.
[9]Warren JD,Blumbergs PC,Thompson PD.Rhabdomyolysis:a review[J].Muscle & Nerve ,2002,25(3):332 -347.
