RRM2B基因突變致線粒體DNA耗竭綜合征:兩例不同類型(8A和8B)患兒的臨床特點和基因分析
2024-07-11 10:19:33鄧琳逯軍
中國全科醫學
2024年29期
鄧琳 逯軍
基金項目:海南省重點研發計劃項目(ZDYF2021SHFZ241)
引用本文:鄧琳,逯軍. RRM2B基因突變致線粒體DNA耗竭綜合征:兩例不同類型(8A和8B)患兒的臨床特點和基因分析[J]. 中國全科醫學,2024,27(29):3704-3708. DOI:10.12114/j.issn.1007-9572.2023.0520. [www.chinagp.net]
DENG L,LU J. Mitochondrial DNA depletion syndrome caused by RRM2B gene mutation:clinical characteristics and genetic analysis of two cases with different types(8A and 8B)[J]. Chinese General Practice,2024,27(29):3704-3708.
? Editorial Office of Chinese General Practice. This is an open access article under the CC BY-NC-ND 4.0 license.
【摘要】 RRM2B基因突變相關疾病根據遺傳方式和臨床表型可分為線粒體DNA耗竭綜合征8A型(MTDPS8A),線粒體DNA耗竭綜合征8B型(MTDPS8B),錐桿營養不良、感音神經性耳聾和范可尼型腎功能障礙(RCDFRD),常染色體顯性進行性眼外肌麻痹伴線粒體DNA缺失5型(PEOA5)這4種類型。其中MTDPS8A、MTDPS8B均屬于線粒體DNA耗竭綜合征,遺傳方式相同,在疾病早期臨床表型復雜且具有異質性,難以鑒別。本文通過系統回顧分析2例分別確診MTDPS8A、MTDPS8B患兒的臨床特點、基因檢測結果、診治經過等病例資料,并復習相關文獻來總結這兩型的遺傳學特點,為今后遇到疑似病例提供診斷思路,進一步提高RRM2B基因突變相關線粒體腦肌病的臨床診斷率,也有助于評估預后情況。
【關鍵詞】 RRM2B基因;線粒體DNA耗竭綜合征8A型;線粒體DNA耗竭綜合征8B型;基因檢測;全外顯子測序
【中圖分類號】 R 725.9 【文獻標識碼】 D DOI:10.12114/j.issn.1007-9572.2023.0520
Mitochondrial DNA Depletion Syndrome Caused by RRM2B Gene Mutation:Clinical Characteristics and Genetic Analysis of Two Cases with Different Types(8A and 8B)
DENG Lin1,LU Jun1,2*
1.Department of Pediatrics,Central South University Xiangya School of Medicine Affiliated Haikou Hospital,Haikou 570208,China
2.Department of Pediatrics,Affiliated Hospital of Guangdong Medical University,Zhanjiang 524000,China
*Corresponding author:LU Jun,Professor/Chief physician/Doctoral supervisor;E-mail:Lu139762@163.com
【Abstract】 RRM2B gene mutation-related diseases can be divided into four types according to genetic pattern and clinical phenotype of mitochondrial DNA depletion syndrome 8A(MTDPS8A),mitochondrial DNA depletion syndrome 8B(MTDPS8B),rod-cone dystrophy,sensorineural deafness,and fanconi-type renal dysfunction(RCDFRD),progressive external ophthalmoplegia with mitochondrial and deletions,autosomal dominant 5(PEOA5). Among them,MTDPS8A and MTDPS8B are both mitochondrial DNA depletion syndromes with the same genetic pattern,and the clinical phenotypes are complex and heterogeneous in the early stage,making it difficult to identify them. This paper systematically reviewed and analyzed the clinical characteristics,genetic test results,diagnosis and treatment process and other case data of two children diagnosed with MTDPS8A and MTDPS8B,and reviewed relevant literature to summarize the genetic characteristics of these two types,so as to provide diagnostic ideas for future suspected cases and further improve the clinical diagnosis rate of RRM2B mutation-related mitochondrial encephalomyopathy,which also helps to assess the prognosis.
【Key words】 RRM2B gene;Mitochondrial DNA depletion syndrome 8A;Mitochondrial DNA depletion syndrome 8B;Genetic testing;Whole-exome sequencing
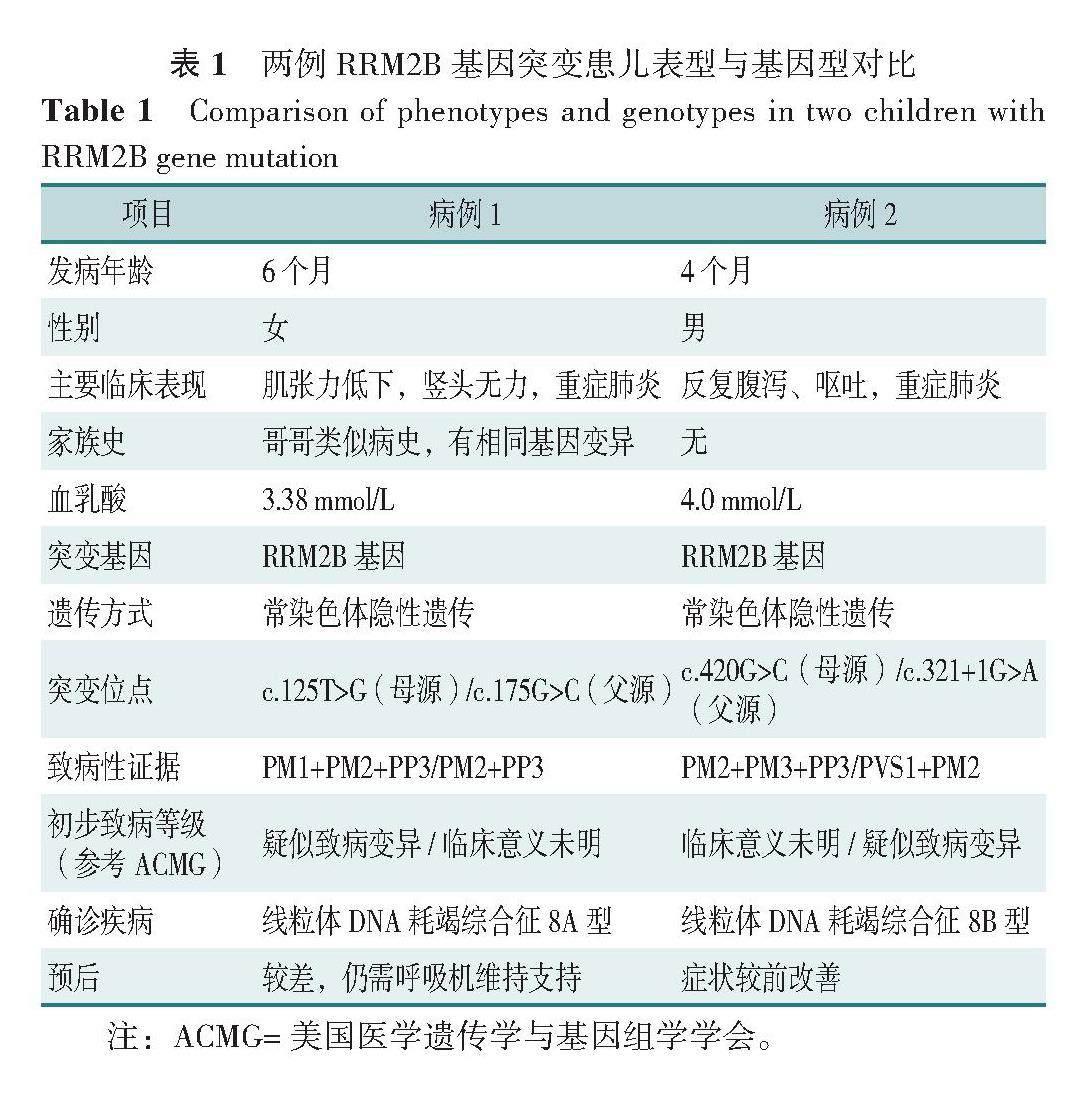
RRM2B基因突變可導致多種遺傳性線粒體疾病,目前已報道的疾病類型包括線粒體DNA耗竭綜合征8A型(mitochondrial DNA depletion syndrome 8A,MTDPS8A),線粒體DNA耗竭綜合征8B型(mitochondrial DNA depletion syndrome 8B,MTDPS8B),錐桿營養不良、感音神經性耳聾和范可尼型腎功能障礙(rod-cone dystrophy,sensorineural deafness,and fanconi-type renal dysfunction,RCDFRD),常染色體顯性進行性眼外肌麻痹伴線粒體DNA缺失5型(progressive external ophthalmoplegia with mitochondrial and deletions,autosomal dominant 5,PEOA5)[1-2]。RRM2B基因突變所致的綜合征中MTDPS8A、MTDPS8B、RCDFRD為常染色體隱性遺傳,而PEOA5為常染色體顯性遺傳,根據遺傳方式可初步鑒別各類綜合征[3]。本文通過描述2例攜帶RRM2B相同致病基因而分別出現MTDPS8A及MTDPS8B兩種不同臨床表型的患兒臨床資料,分析表型與基因型的相關性,以期提高臨床醫師對RRM2B基因突變相關疾病不同分型遺傳學的認識及診治水平。……
登錄APP查看全文
