歸芪通脈顆粒的質量標準研究
2022-06-01 07:17:46吳作敏于曉濤陳恒文金少舉
食品與藥品 2022年3期
郭 麗,陳 皓,吳作敏,于曉濤,陳恒文,金少舉,王 瑞*
(1. 漯河市第一人民醫院 藥學部,河南 漯河 462300;2. 中國中醫科學院廣安門醫院 心血管科,北京 100053;3. 漯河醫學高等專科學校 藥理學教研室,河南 漯河 462002)
歸芪通脈方是由黃芪、當歸、黨參、陳皮、蜈蚣等14味藥材經科學配伍組成的醫院制劑,具有補氣活血,化瘀通絡之功效,用于改善缺血性腦卒中恢復期的言語謇澀,肢體麻木等,在我院臨床療效良好,具有開發成為名優中成藥的潛力和價值。方中君藥黃芪的主要活性成分黃芪甲苷,通過抑制內皮細胞凋亡,促進內皮細胞的增殖和遷移等,對缺血缺氧性腦血管內皮損傷起到保護作用[1];芍藥苷是臣藥赤芍活血化瘀的功效物質基礎[2],有研究芍藥苷通過PC12細胞中的Bcl-2/Bax通路防止谷氨酸誘導的神經毒性發揮神經保護作用[3];黨參炔苷是佐藥黨參質量評價的指標性成分之一[4]。
歸芪通脈方經提取優化工藝制備而成[5],為了便于患者攜帶和服用,通過提取濃縮而后經噴霧干燥開發成為顆粒劑。為了對該制劑進行質量控制,本研究采用薄層色譜法對處方中黃芪、赤芍、黨參進行定性鑒別,采用HPLC-DAD雙波長檢測法對羥基紅花黃色素A(HSYA,紅花)、毛蕊異黃酮葡萄糖苷(黃芪)、芍藥苷(赤芍)和橙皮苷(陳皮)4種有效成分進行含量測定[6],為開發治療中風中藥新藥提供理論依據。
1 儀器與材料
1.1 儀器
Shimadzu LC 20AD型HPLC儀(二級管陣列檢測器);AUW-120D型電子天平(精度為十萬分之一,日本Shimadzu公司);SB25-12D超聲波清洗機(寧波新芝生物科技股份有限公司);DHG-9070型電熱鼓風干燥箱(上海一恒科學儀器有限公司)。
1.2 試藥
對照品HSYA(批號:MUST-19082111,純度:98.11 %)、橙皮苷(批號:MUST-19030701,純度:98.46 %)均購自成都曼思特生物科技有限公司,對照品黃芪甲苷(批號:110781-201616,純度97.40 %)、毛蕊異黃酮葡萄糖苷(批號:111920-201606,含量:97.60 %)、芍藥苷(批號:110736-201842,含量:97.40 %),對照藥材黨參(批號:121057-201206)均購自中國食品藥品檢定研究院;乙腈為色譜純;三氯甲烷、乙酸乙酯等試劑為分析純,水為超純水。供試品歸芪通脈顆粒(批號:20200426,20200529,20200626)及陰性樣品,由漯河市第一人民醫院制劑室提供。
2 方法與結果
2.1 TLC鑒別
2.1.1 黃芪TLC鑒別[7-8]精密稱取歸芪通脈顆粒10 g,加水20 ml使其溶解,水飽和的正丁醇提取3次,每次30 ml,合并正丁醇液,用氨試液洗滌3次,每次20 ml,再用水洗至中性,用無水硫酸鈉脫水,正丁醇液蒸干,殘渣加甲醇1 ml溶解,作為供試品溶液;同法制成陰性樣品溶液。另取黃芪甲苷對照品適量,加甲醇制成每1 ml含1 mg的對照品溶液。照薄層色譜法(通則0502)試驗,吸取上述3種溶液各5 μl,分別點于同一硅膠G薄層板上,以三氯甲烷-甲醇-水(13:7:2)10 ℃以下放置的下層溶液為展開劑,展開,取出,晾干,噴以10 %硫酸乙醇溶液,105 ℃加熱至斑點顯色清晰。供試品色譜中,在與對照品色譜相應的位置上,日光下顯相同顏色的棕褐色斑點;紫外光下顯相同顏色的橙黃色熒光斑點,陰性樣品在相同位置上無干擾,結果見圖1。
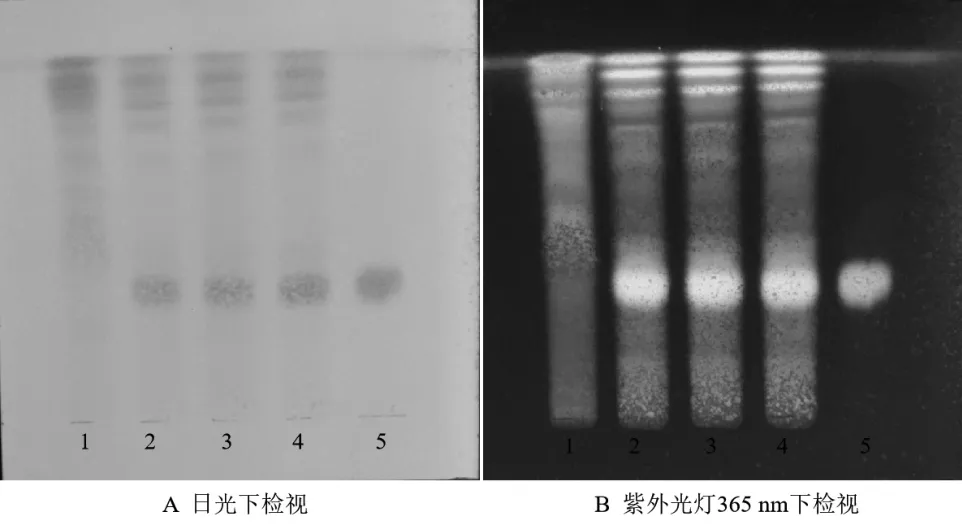
圖1 黃芪TLC鑒別
2.1.2 赤芍TLC鑒別[9]精密稱取歸芪通脈顆粒5 g,加水20 ml使其溶解,用水飽和的正丁醇提取3次,每次20 ml,合并正丁醇液,蒸干,殘渣加甲醇1 ml溶解,作為供試品溶液;同法制成陰性樣品溶液。另取芍藥苷對照品,加甲醇制成每1 ml含1 mg的溶液,作為對照品溶液。照薄層色譜法(通則0502)進行試驗,吸取上述3種溶液各4 μl,分別點于同一硅膠G薄層板上,以三氯甲烷-乙酸乙酯-甲醇-甲酸(40:5:10:0.2)為展開劑,展開,取出,晾干,噴以5 %香草醛硫酸溶液,105 ℃加熱至斑點顯色清晰。供試品色譜中,在與對照品色譜相應的位置上,顯相同顏色的斑點,陰性樣品在相應位置上無干擾,結果見圖2。

圖2 赤芍TLC鑒別
2.1.3 黨參TLC鑒別[4]精密稱取歸芪通脈顆粒10 g,加水20 ml,置圓底燒瓶中,加鹽酸3 ml,加熱回流30 min,放冷,濾過,濾液用石油醚(30~60 ℃)提取2次,每次40 ml,棄去石油醚液,再用二氯甲烷提取3次,每次20 ml,合并二氯甲烷提取液,殘渣加二氯甲烷1 ml使溶解,作為供試品溶液;同法制成陰性樣品溶液。另取黨參對照藥材1 g,加水適量,煎煮1 h,放冷,濾過,濾液濃縮至約20 ml,同法制成對照藥材溶液。照薄層色譜法(通則0502)試驗,吸取上述3種溶液各4 μl,分別點于同一硅膠G薄層板上,以甲苯-乙酸乙酯-甲酸(20:18:0.5)為展開劑,展開,取出,晾干,噴以10 %硫酸乙醇溶液,105 ℃加熱至斑點顯色清晰。供試品色譜中,在與對照品色譜相應的位置上,顯相同顏色的深褐色斑點,陰性樣品無干擾,結果見圖3。
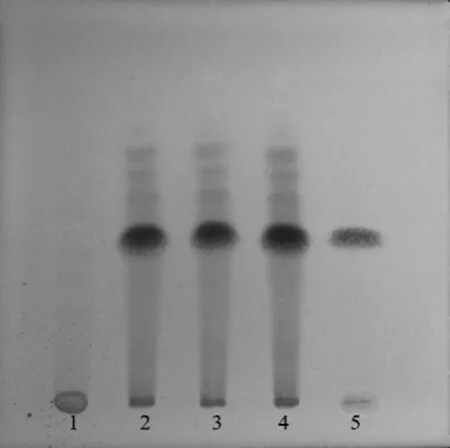
圖3 黨參TLC鑒別
2.2 含量測定
2.2.1 色譜條件 色譜柱為Gemini Phenomenex C18(250 mm×4.6 μm,5 μm);流動相為乙腈(A)-0.1%磷酸水溶液(B),梯度洗脫(0~5 min,15 %→20 % A;5~10 min,20 %→22 % A;10~20 min,22 % A;20~40 min,22 %→80 % A;40~45 min,80 % A;45~48 min,15 % A)。經PDA全波長掃描,確定最佳檢測波長為403 nm(HSYA)、220 nm(芍藥苷、毛蕊異黃酮葡萄糖苷、橙皮苷),故采用雙波長檢測。流速為0.8 ml/min,柱溫為25 ℃,進樣量為5 μl。
2.2.2 溶液的制備
2.2.2.1 對照品溶液制備 精密稱取對照品HSYA、芍藥苷、毛蕊異黃酮葡萄糖苷、橙皮苷各適量,置入10 ml量瓶,加甲醇溶解并定容,配成質量濃度分別為255.10,146.10,116.10,204.80 μg/ml的混合對照品儲備液。
2.2.2.2 供試品溶液制備 精密稱取歸芪通脈顆粒(批號:20200426)2.50 g,研細,置入25 ml量瓶,用70 %乙醇稀釋至刻度,稱重,超聲30 min,放冷稱重并補足失重,搖勻,濾過,取續濾液10 ml蒸干,殘渣加70 %乙醇使溶解,轉移至10 ml量瓶,作為供試品溶液。
2.2.2.3 陰性樣品溶液制備 分別取缺黃芪、缺赤芍、缺紅花、缺陳皮的陰性樣品,按供試品溶液制備方法制成陰性樣品溶液。
2.2.3 專屬性考察 分別精密吸取2.2.2項下制備的混合對照品溶液、供試品溶液、陰性樣品溶液各適量,按2.2.1項下色譜條件進樣測定,見圖4。結果表明,供試品中HSYA、芍藥苷、毛蕊異黃酮葡萄糖苷和橙皮苷與其他組分的分離度大于1.5,與對照品色譜在相應的位置上具有相同的保留時間,陰性樣品溶液對這4種成分的測定均無干擾。
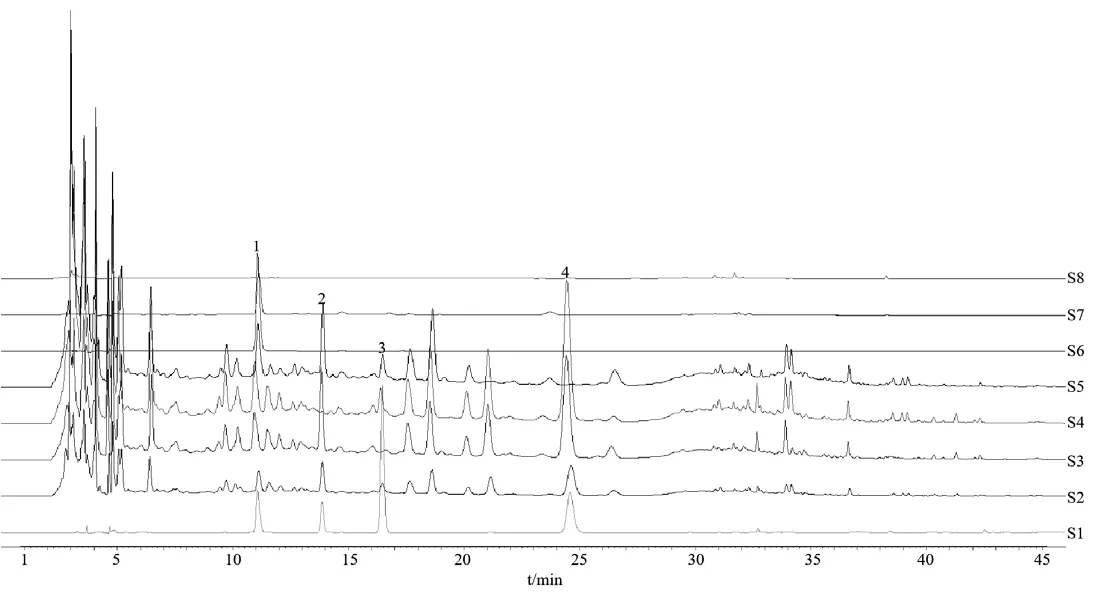
圖4 歸芪通脈顆粒高效液相色譜圖
2.2.4 線性關系考察 精密吸取混合對照品儲備液2.5 ml,置入5 ml量瓶,用甲醇稀釋至刻度,逐級稀釋,制成6個濃度梯度的混合對照品溶液,依法進樣測定,以質量濃度(μg/ml)為橫坐標(x),4種成分的峰面積(y)為縱坐標進行線性回歸,結果見表1。
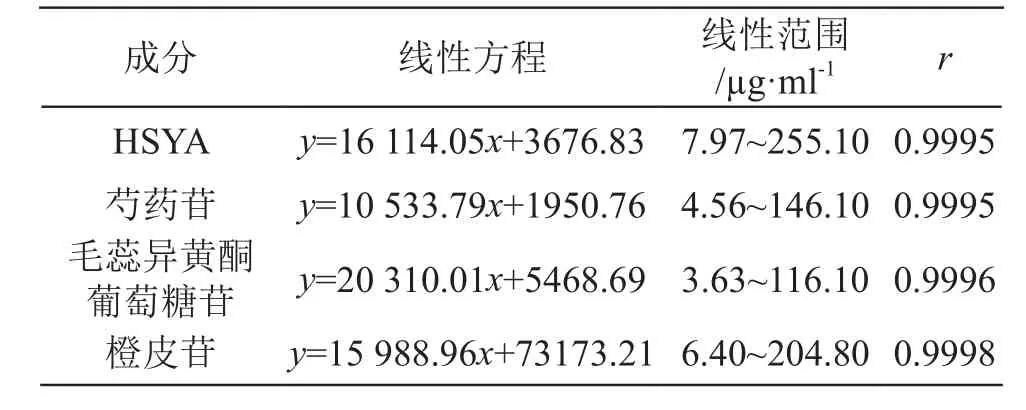
表1 線性關系考察結果
2.2.5 精密度試驗 精密吸取2.2.2項下的混合對照品溶液,連續進樣6次,按照2.2.1項色譜條件測定并記錄峰面積。結果,4種成分峰面積的RSD分別為0.29 %,0.21 %,0.31 %,1.74 %(n=6),表明儀器精密度良好。
2.2.6 重復性試驗 取同一批樣品(批號:20200426)2.50 g,按2.2.2項下方法平行制備6份供試品溶液,按2.2.1項色譜條件分別測定峰面積。結果,4種成分峰面積的RSD分別為1.09 %,1.80 %,2.51 %,2.90 %(n=6),表明重復性良好。
2.2.7 穩定性試驗 取2.2.6項下同一供試品溶液,于室溫下放置0,2,4,8,12,24 h分別進樣測定,記錄峰面積。結果4種成分峰面積的RSD分別為1.17 %,1.07 %,2.14 %,2.98 %(n=6),表明24 h內供試品溶液穩定性良好。
2.2.8 回收率試驗 取同一批次已知含量的歸芪通脈顆粒適量,研細,精密稱取1.25 g,共6份,分別置入25 ml量瓶,加入一定質量的混合對照品,加甲醇稀釋至刻度,搖勻,濾過,取續濾液,制得供試品溶液,計算回收率及RSD,見表2~5。結果,HSYA、芍藥苷、毛蕊異黃酮葡萄糖苷、橙皮苷的平均回收率分別為101.04 %,98.31 %,99.33 %,98.79 %,RSD分別為1.74 %,1.12 %,1.36 %,1.51 %。表明該方法的回收率良好。
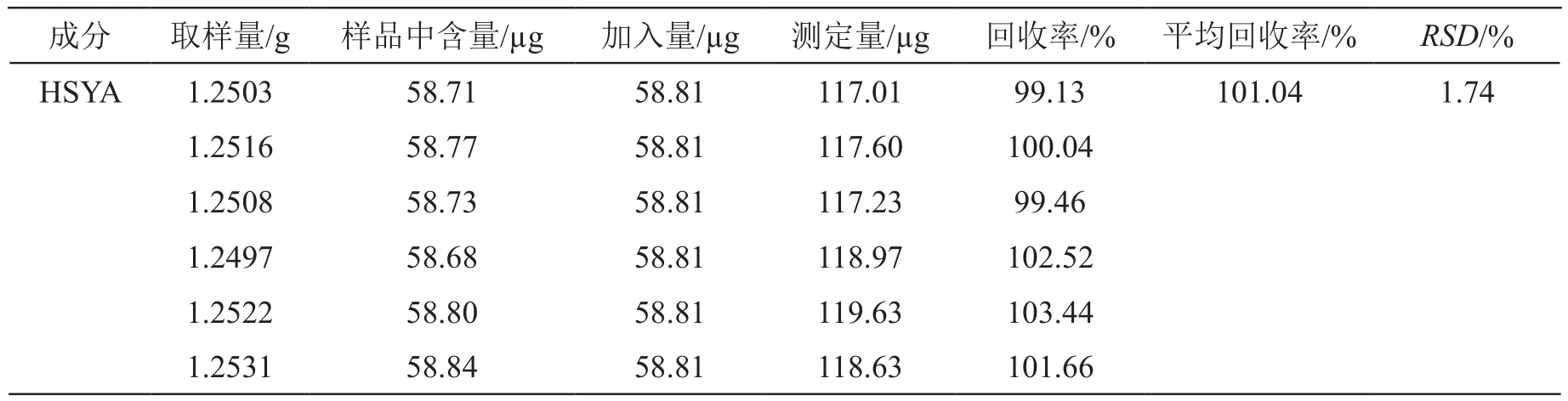
表2 HSYA加樣回收率結果(n=6)
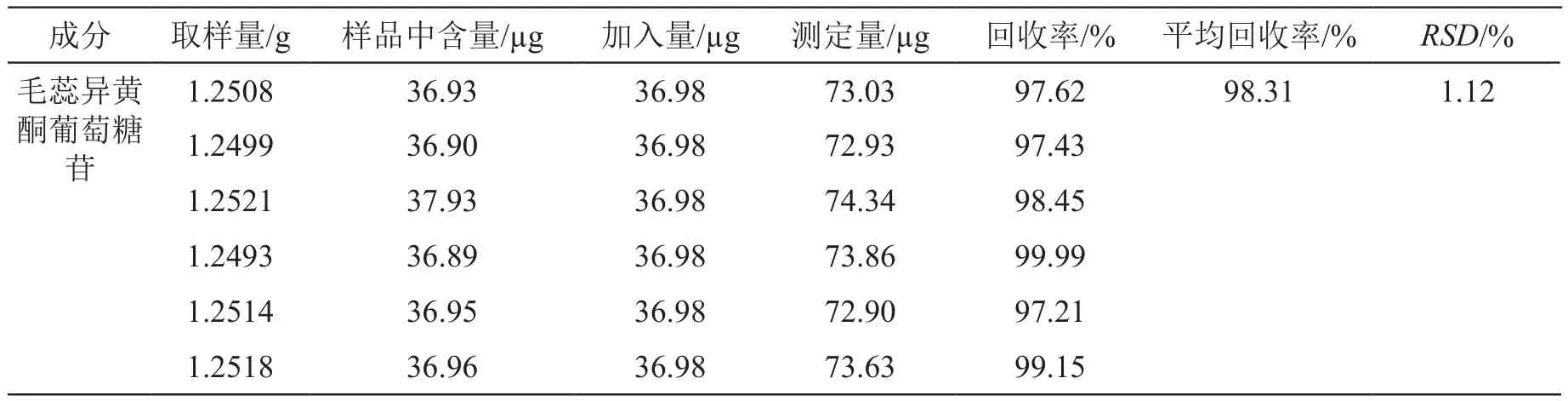
表3 毛蕊異黃酮葡萄糖苷加樣回收率結果(n=6)
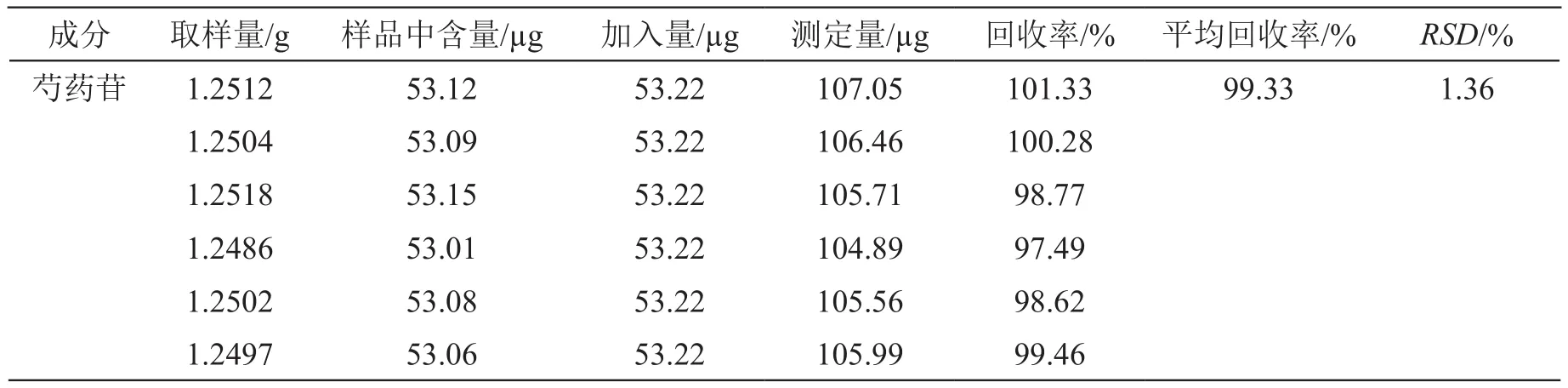
表4 芍藥苷加樣回收率結果(n=6)
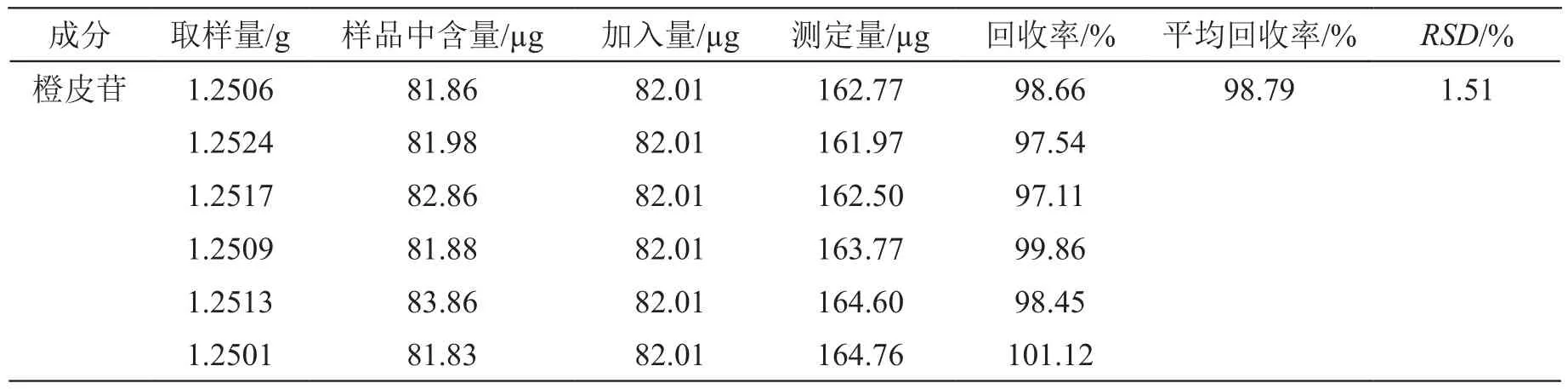
表5 橙皮苷加樣回收率結果(n=6)
2.3 樣品的含量測定
取10批歸芪通脈顆粒,按2.2.2項下方法制備供試品溶液,按2.2.1項下色譜條件進樣,見表6。將所測各成分含量平均值的80 %作為內控指標,暫將含量限度定為:每1 g含紅花以羥甲基紅花黃色素A(C27H32O16)計不少于0.07 mg,含芍藥以芍藥苷(C23H28O11)計不少于0.06 mg,含黃芪以毛蕊異黃酮葡萄糖苷(C22H22O10)計不少于0.04 mg,含陳皮以橙皮苷(C28H34O15)計不少于0.10 mg。
3 討論
3.1 TLC鑒別方法優化
在鑒別黨參時先借鑒了黃芪項下的樣品制備方法,所得TLC色譜圖陰性有干擾且無明顯特征點,后將樣品酸化,置沸水浴,用二氯甲烷水解得到苷元,得到了專屬性強且陰性無干擾的黨參TLC色譜圖。
當歸、川芎中均含有阿魏酸,具有抗血栓形成和抑制血小板聚集等作用[10-11]。實驗中曾嘗試同時對當歸、川芎進行薄層鑒別,但發現供試品與對照藥材分別對應有明顯的亮斑點,但缺當歸和川芎的陰性對照溶液卻存在干擾,又經過HPLC色譜的對比發現在標準品阿魏酸的相同保留時間和位置存在干擾峰,表明當歸和川芎的薄層鑒別不具有專屬性和特征性,故未列入標準。曾嘗試白術的薄層鑒別,但發現陰性有干擾,故未列入標準。本研究建立的黃芪、芍藥、黨參的薄層鑒別方法重現性好,分離效果好,特征斑點清晰且陰性無干擾,能有效控制該制劑的質量。
3.2 HPLC含量測定方法優化
含量測定的指標選擇了君、臣、佐藥,首先采用HPLC-ELSD對君藥黃芪甲苷進行含量測定,發現樣品制備操作復雜,誤差較大,故未列入標準。為減少誤差,選用70 %乙醇直接稀釋樣品,離心取上清的方法制備樣品,同時建立HSYA、芍藥苷、毛蕊異黃酮葡萄糖苷和橙皮苷4個含量測定,采用雙波長法進行含量測定,HSYA(403 nm),芍藥苷、毛蕊異黃酮葡萄糖苷和橙皮苷(220 nm),并且經過方法學考察,專屬性強,精密度高,重現性好。
綜上所述,本研究建立歸芪通脈顆粒的定性定量方法,符合中藥質量標準制定的技術要求,可作為該制劑的質量控制標準。
