一測多評法同時測定土茯苓配方顆粒中5種成分的含量
2022-04-26 07:53:38何榮榮嚴玉晶王瑜婷田清清徐東婷余欣彤魏梅
廣東藥科大學學報 2022年2期
關鍵詞:質量
何榮榮,嚴玉晶,王瑜婷,田清清,徐東婷,余欣彤,魏梅
(廣東一方制藥有限公司/廣東省中藥配方顆粒企業重點實驗室,廣東佛山528244)
土茯苓為百合科植物光葉菝葜Smilax glabraRoxb.的干燥根莖,始載于《本草經集注》,別稱禹余糧、過山龍、刺豬苓等,其味甘、淡,性平,歸肝、胃經,具有解毒、除濕、通利關節的功效[1]。土茯苓主要含有黃酮類、皂苷類、生物堿類、有機酸類、苯丙素類、鞣質等多種有效成分,是治療痛風的傳統中藥,具有抗炎、抗氧化、抗腫瘤、保護肝損傷、免疫調節、利尿、抑菌等諸多藥理活性[2?6]。
國家藥品監督管理局頒發的土茯苓配方顆粒質量標準含量測定項下只規定了落新婦苷的含量[7],而中藥成分復雜多樣,單一指標成分的定量分析模式,難以全面反映土茯苓配方顆粒的質量,不能滿足中藥現代化對質量控制的要求。一測多評法(quantitative analysis of multi?components by single?maker,QAMS)是根據中藥有效成分內在函數關系和比例關系,只測定一個成分(對照品易得、穩定者)即可實現多個成分(對照品匱乏者)的同步測定[8],是一種用于多成分質量控制的新模式。本研究以相對價廉、易得的落新婦苷為內標物,通過相對校正因子(relative correction fator,RCF)計算各待測成分的質量分數,并與外標法(esternal standard method,ESM)測定結果進行對比,考察QAMS 在土茯苓配方顆粒多指標成分質量評價中的準確性、適用性,以期為土茯苓配方顆粒的質量控制提供參考。
1 儀器與材料
1.1 儀器
Waters H?CLASS 型超高效液相色譜儀(美國Waters公司);Agilent 1290型超高效液相色譜儀(美國Agilent公司);XP26型百萬分之一天平、ME204E型萬分之一天平(瑞士METTLER TDLEDO 公司);Milli?Q Direct 型超純水系統(德國Merck 公司);KQ?500DE 型數控超聲波清洗器(昆山市超聲儀器有限公司)。
1.2 試藥
新落新婦苷(批號:DSTDX007701,純度≥98%)、新異落新婦苷(批號:DXTDX021601,純度≥98%)、異落新婦苷(批號:DXTDX007801,純度≥95%)對照品均購自成都德思特生物技術有限公司;落新婦苷(批號:111798?201805,純度93.6%)、黃杞苷(批號:111906?201102,純度93.7%)對照品均購自中國食品藥品檢定研究院;甲醇、乙腈為色譜純(德國Merck公司);乙酸為色譜純(天津市科密歐化學試劑有限公司);水為超純水,其他試劑為分析純。10 批土茯苓配方顆粒(編號S1-S10)由廣東一方制藥有限公司制備,具體批號見表1。

表1 樣品信息表Table 1 Sample information table
2 方法與結果
2.1 色譜條件[7]
色譜柱:Agilent ZORBAX SB?C18(100 mm×2.1 mm,1.8 μm);流動相:乙腈(A)?0.2%乙酸溶液(B)梯度洗脫(0~2 min,13%A;2~5 min,13%~17%A;5~14 min,17%~23%A);檢測波長:291 nm;柱溫:30 ℃;流速:0.4 mL/min;進樣量:1 μL。
2.2 混合對照品溶液的制備
分別取新落新婦苷、落新婦苷、新異落新婦苷、異落新婦苷、黃杞苷適量,精密稱定,置25 mL 量瓶中,加60%(體積分數,下同)甲醇定容至刻度,制成質量濃度分別為49.705 6、46.949 8、48.222 0、49.666 4、47.149 8 μg/mL的混合對照品溶液。
2.3 供試品溶液的制備
取土茯苓配方顆粒適量,研細,取約0.1 g,精密稱定,置具塞錐形瓶中,精密加入70%(體積分數,下同)乙醇100 mL,稱定質量,超聲處理(功率300 W,頻率45 kHz)40 min,放冷,再稱定質量,用70%乙醇補足減失的質量,搖勻,濾過,取續濾液,即得。
2.4 方法學考察
2.4.1 專屬性試驗分別精密吸取空白溶劑、混合對照品溶液及供試品溶液,按“2.1”項下色譜條件進樣測定,結果見圖1。可見,供試品溶液色譜與混合對照品溶液色譜在相應的保留時間處具有相同的色譜峰,且空白溶劑無干擾,表明方法專屬性良好。

圖1 專屬性試驗UPLC色譜圖Figure 1 UPLC chromatograms of specificity test
2.4.2 線性關系考察分別精密吸取“2.2”項下混合對照品溶液0.2、1、2、4、6、8 mL 置10 mL 量瓶中,加60%甲醇定容至刻度,得線性混合對照品溶液。按“2.1”項下色譜條件進樣測定,記錄色譜圖。以對照品溶液質量濃度為橫坐標(X),峰面積為縱坐標(Y)繪制標準曲線,結果見表2,表明各成分在各自濃度范圍內線性關系良好。
2.4.3 精密度試驗精密吸取“2.2”項下混合對照品溶液,按“2.1”項下色譜條件連續進樣6 次,計算新落新婦苷、落新婦苷、新異落新婦苷、異落新婦苷、黃杞苷峰面積的RSD 值分別為0.05%、0.22%、0.13%、0.18%、0.12%,表明儀器精密度良好。
2.4.4 重復性試驗取土茯苓配方顆粒(編號S1)適量,按“2.3”項下方法平行制備6 份供試品溶液,按“2.1”項下色譜條件進樣測定,計算新落新婦苷、落新婦苷、新異落新婦苷、異落新婦苷、黃杞苷的平均質量分數分別為1.74%、1.30%、0.84%、0.51%、0.11%,其RSD 值分別為1.33%、1.46%、1.55%、1.34%、2.34%,表明方法重復性良好。
2.4.5 穩定性試驗取土茯苓配方顆粒(編號S1)適量,按“2.3”項下方法制備供試品溶液,按“2.1”項下色譜條件分別在0、2、4、6、8、12 h 進樣測定,計算新落新婦苷、落新婦苷、新異落新婦苷、異落新婦苷、黃杞苷峰面積的RSD 值分別為0.57%、0.68%、0.34%、0.61%、1.41%,表明供試品溶液在12 h 內穩定性良好。
2.4.6 加樣回收率試驗取已知質量分數的土茯苓配方顆粒(編號S1)適量,研細,精密稱取9 份,每份約0.05 g,置具塞錐形瓶中,分為3組,分別精密加入相當于供試品中各被測成分質量分數50%、100%、150%的混合對照品溶液(新落新婦苷、落新婦苷、新異落新婦苷、異落新婦苷、黃杞苷質量濃度分別為52.420 2、61.177 0、40.935 5、23.373 0、6.156 1 μg/mL),揮干溶劑,按“2.3”項下方法制備供試品溶液,按“2.1”項下色譜條件進樣測定,計算加樣回收率和RSD,結果見表3,表明該方法準確性良好。
2.5 相對校正因子(RCF)的確定[9?10]
分別精密吸取“2.2”項下混合對照品溶液0.2、0.5、0.8、1.0、1.5、2.0 μL,按“2.1”項下色譜條件進樣測定,以落新婦苷為內標物,按公式fj/s=fj/fs=(Aj*Cs)/(As*Cj)(As、Aj分別為內標物和待測成分峰面積,Cs、Cj分別為內標物和待測成分質量濃度)計算新落新婦苷、新異落新婦苷、異落新婦苷、黃杞苷的RCF,結果見表4。
2.6 耐用性和系統適應性研究[11?12]
2.6.1 不同儀器及色譜柱對RCF 的影響以落新婦苷為內標物,考察其他4 種成分在Waters H?CLASS、Agilent 1290 型2 種超高效液相色譜系統,Waters HSS T3(100 mm×2.1 mm,1.8 μm)、Waters CORTECS T3(100 mm×2.1 mm,1.8 μm)、Agilent ZORBAX SB?C18(100 mm×2.1 mm,1.8 μm)3 種色譜柱下的RCF,結果見表5。可見,各成分RCF 的RSD 均小于3%,表明不同的儀器和色譜柱對各成分RCF無顯著影響。
2.6.2 不同柱溫對RCF的影響采用Waters H?CLASS型超高效液相色譜儀、Agilent ZORBAX SB?C18柱(100 mm×2.1 mm,1.8 μm),考察3 種不同柱溫(25、30、35 ℃)對各成分RCF 的影響,結果見表6。可見,各成分RCF的RSD 均小于3%,表明柱溫的變化對各成分RCF無顯著影響。
2.6.3 不同流速對相對校正因子的影響采用Waters H?CLASS 型超高效液相色譜儀、Agilent ZORBAX SB?C18柱(100 mm×2.1 mm,1.8 μm),考察3 種不同流速(0.2、0.3、0.4 mL/min)對各成分RCF的影響,結果見表7。可見,各成分RCF 的RSD 均小于3%,表明流速的變化對各成分RCF無顯著影響。

表2 線性關系考察結果Table 2 Results of linear relationship
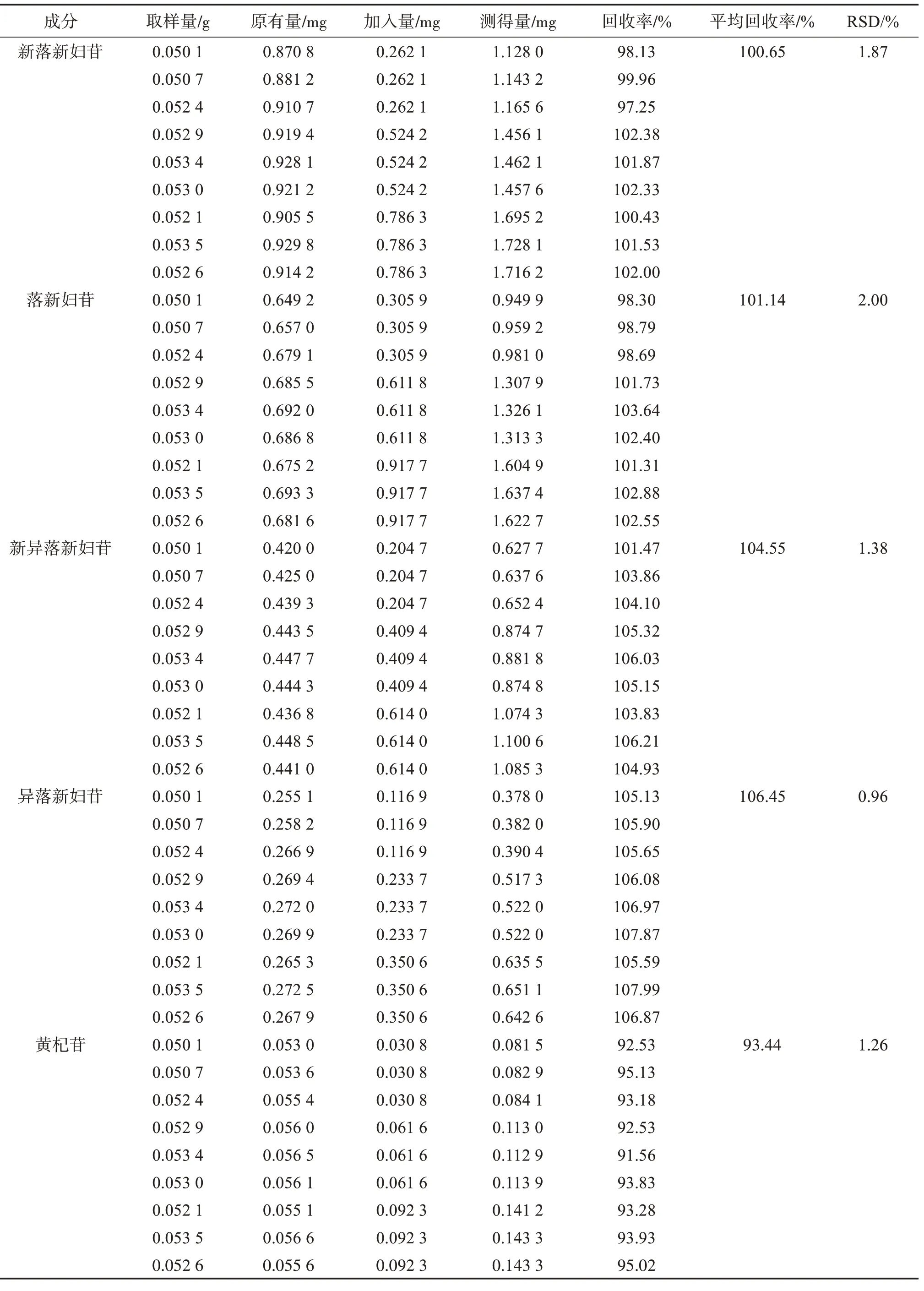
表3 土茯苓配方顆粒中5種成分的加樣回收率試驗結果Table 3 Sample recovery test results of 5 components inSmilax glabraformula granules
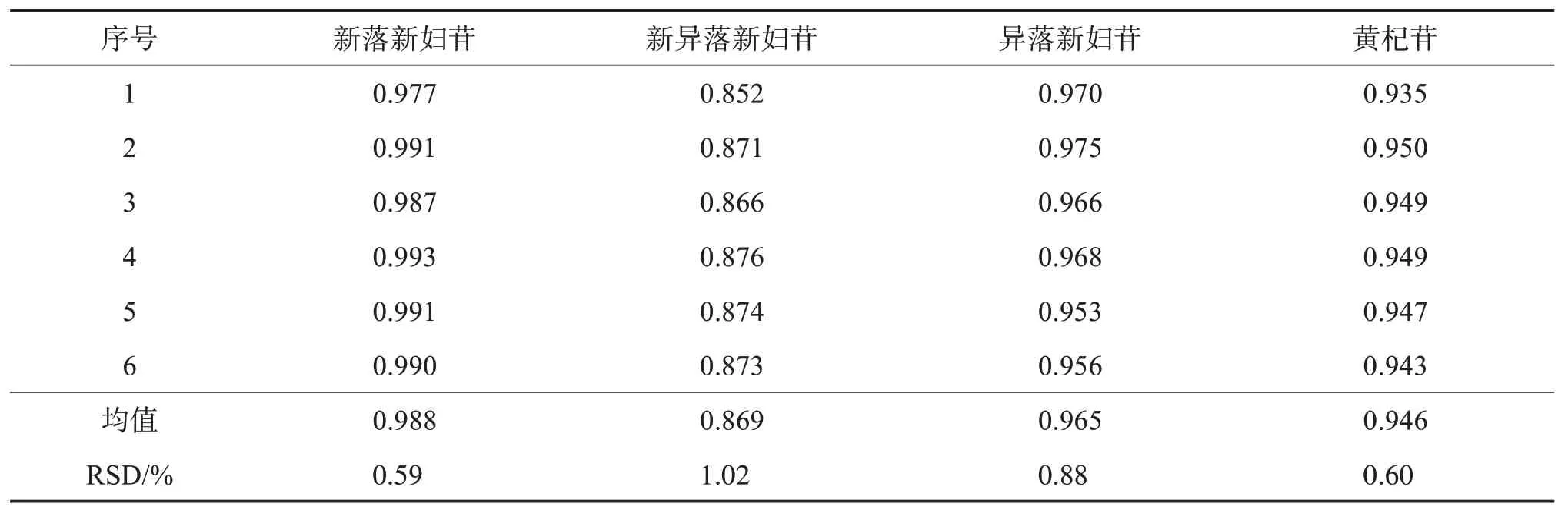
表4 各成分的RCFTable 4 RCF of various constituents

表5 不同儀器、色譜柱下各成分的RCFTable 5 Effects of different instruments and columns on RCF
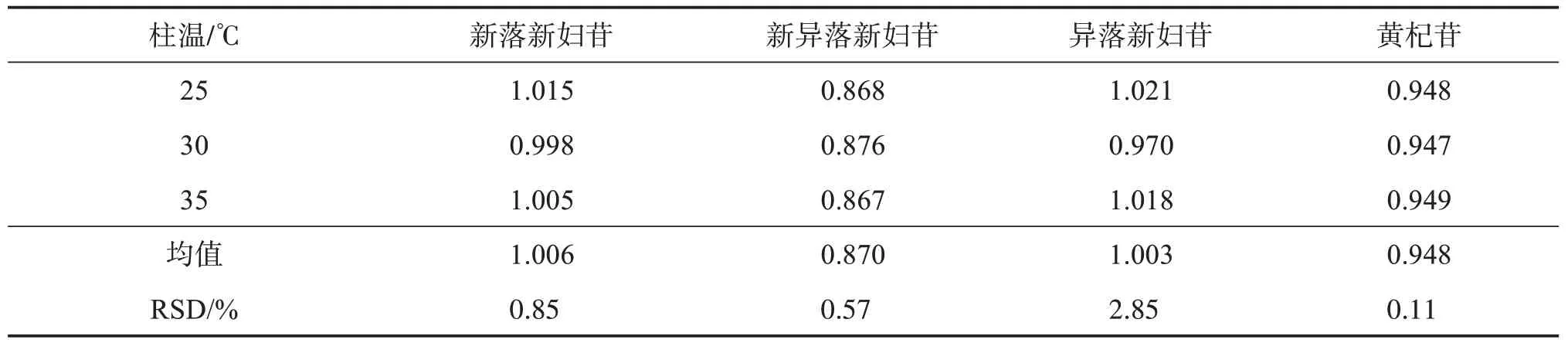
表6 不同柱溫下各成分的RCFTable 6 Effects of different column temperature on RCF

表7 不同流速下各成分的RCFTable 7 Effects of different flow rates on RCF
2.7 待測成分色譜峰的定位
一測多評法可利用相對保留時間法、保留時間差法來準確定位各待測成分色譜峰[13?14]。本研究以落新婦苷為內標物,考察其他4 種成分在Waters H?CLASS、Agilent 1290 型2 種超高效液相色譜系統,Waters HSS T3(100 mm×2.1 mm,1.8 μm)、Wa?ters CORTECS T3(100 mm×2.1 mm,1.8 μm)、Agi?lent ZORBAX SB?C18(100 mm×2.1 mm,1.8 μm)3 種色譜柱下的相對保留時間,以此增加各定位組分的可信度,結果見表8。可見,各成分相對保留時間的RSD 均小于3%,表明采用相對保留時間進行色譜峰定位是可行的。

表8 各成分的相對保留時間Table 8 Relative retention time of various constituents
2.8 QAMS與ESM比較
取10 批土茯苓配方顆粒,按“2.3”項方法制備供試品溶液,按“2.1”項下色譜條件進樣測定,運用SPSS20.0 軟件對ESM 實測值和QAMS 計算結果進行成對t檢驗及Pearson相關系數(r)分析,結果見表9。可見,2 種方法之間的相關系數r均為1.000,t檢驗顯示差異無統計學意義(P>0.05),表明建立的QAMS準確、可行。

表9 ESM與QAMS測定結果比較Table 9 Comparison of results obtained by ESM and QAMS method w/(g·100 mg-1)
3 討論
中藥成分的復雜性和多效性致使單一指標成分的定量測定難以體現其整體質量,建立多成分質量分數測定方法能更全面地反映其內在質量。傳統的ESM 進行多成分檢測時需用到多種對照品,且有些中藥化學對照品價格昂貴、不易獲得,QAMS只需1 種對照品即可計算其他多個成分的質量分數,可以有效解決對照品稀缺問題,節約檢驗成本、提高檢測效率。
土茯苓的主要有效成分為黃酮類化合物,包括黃酮醇類、二氫黃酮類、異黃酮類和黃烷醇類等,這些成分與其抗炎、抗癌、抑菌等藥理作用密切相關[15]。已有將QAMS應用于土茯苓藥材中5種黃酮類成分含量測定的報道,結果表明建立的方法穩定可行[16]。中藥配方顆粒是由單味中藥飲片經水提、濃縮、干燥、制粒等工序制得的一種顆粒劑,具有療效確切、服用方便、患者順應性強等優點,具有良好的發展前景,對其產品質量的全面把控也尤為重要。本研究將新落新婦苷、落新婦苷、新異落新婦苷、異落新婦苷、黃杞苷5種黃酮類成分作為土茯苓配方顆粒質量控制的指標,選擇化學性質較穩定且價格適中的落新婦苷為內標物,建立了土茯苓配方顆粒中5 種黃酮類成分的QAMS,確定了4 種成分的RCF。結果表明,不同儀器、色譜柱、柱溫及流速等外部條件對RCF 無顯著影響,且各待測成分色譜峰的相對保留時間均穩定。采用QAMS 與ESM 2種方法測定10 批土茯苓配方顆粒中5 種黃酮類成分的質量分數,所得結果基本一致,說明建立的QAMS 法科學、可靠,可為土茯苓配方顆粒多指標質量控制研究提供參考。
猜你喜歡
中學生數理化·中考版(2022年10期)2022-11-10 09:37:42
中學生數理化·八年級物理人教版(2022年12期)2022-02-14 07:08:42
中學生數理化·八年級物理人教版(2021年12期)2021-12-31 03:23:08
中學生數理化·中考版(2020年10期)2020-11-27 01:59:48
中國生殖健康(2019年2期)2019-08-23 08:12:08
石油化工建設(2018年6期)2018-04-22 03:16:54
產品可靠性報告(2017年7期)2017-09-05 09:49:12
中學生數理化·八年級物理人教版(2017年12期)2017-04-18 12:59:38
汽車觀察(2016年3期)2016-02-28 13:16:26
民生周刊(2014年7期)2014-03-28 01:30:54
