一種改進的Cham-Evans-Lam偶聯反應在1-環丙基-4-碘-1H吡唑制備中的應用
2021-12-21 02:21:20呂志卿陳偉榮張鑫龍
化工時刊 2021年10期
呂志卿 陳偉榮 張鑫龍
(浙江海正藥業股份有限公司,浙江 臺州 318000)
Chan-Evans-Lam 偶聯反應自從1998年被發現以來,已經成為過渡金屬催化氧化交叉偶聯反應用來構建碳-碳鍵、碳-雜鍵最有效的方法之一[1-3],此類反應是在銅鹽促進下,雜原子(N、O或S)作為親核試劑與芳基或是烷基硼酸進行偶聯,從而構建碳-雜鍵。與其他過渡金屬(鈀、鉑、鎳)參與的偶聯反應比,此反應具有催化劑價格便宜,條件溫和,不需要復雜的配體、操作簡便等優點[4],目前,相關的研究報道層出不窮[5,6],反應機理可能如下:
1-環丙基-4-碘-1H吡唑,是一種磷脂酰肌醇3-激酶抑制劑類藥物的關鍵原料,而磷脂酰肌醇3-激酶抑制劑可作為抗癌劑通過激活抗腫瘤免疫反應來攻擊癌癥,因此,該原料若能實現大批量生產對磷脂酰肌醇3-激酶抑制劑類藥物的研究具有重大的意義。以4-碘-1H吡唑和環丙基硼酸為原料,1-環丙基-4-碘-1H吡唑的合成路線見圖2,目前通用的制備方法存在眾多弊端,比如反應溫度高,時間長,同時,所用的試劑存在著重大的EHS風險隱患,工藝可重復性差,收率低等缺點,BOCK、Meng Wei等[7,8]使用二氧六環做溶劑,在吡啶及4-二甲氨基吡啶(DMAP)條件下,在銅鹽的促進下,100 ℃,通氧,反應12 h,可得到60%收率的產物,但是反應中使用到易產生過氧化物二氧六環,反應溫度高,在通氧的條件存在著重大的安全風險,Fujihara等[9]通過使用高絡合性的有機堿性2,2′-聯吡啶,在銅鹽的作用下,加熱反應,但是反應時間長,需要過夜,并且使用1類溶劑1,2-二氯乙烷,此類溶劑具有強烈的致癌性,為工業化生產避免使用的試劑,Miller等[10]對其進行了改進,用1,4-二氧六環代替1,2-二氯乙烷,在加壓的條件下反應,但是收率只有19%。
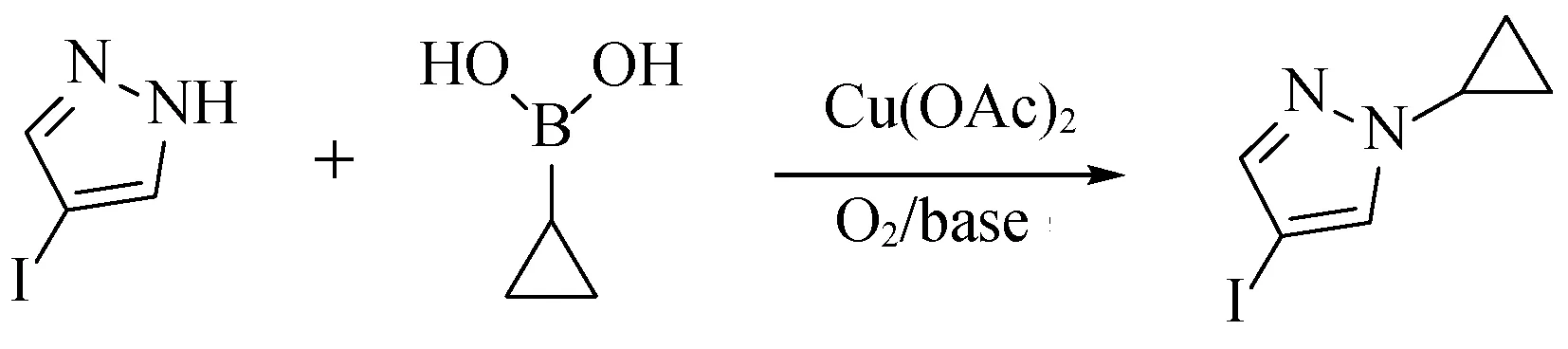
圖2 1-環丙基-4-碘-1H吡唑的合成路線Fig. 2 Synthetic Route of 1-cyclopropyl-4-iodo-1H pyrazole
本文通過對上述反應的條件進行篩選并優化,對相關試劑及溶劑也進行了篩選,最終提出了一種改進后的方法。
1 實驗部分
1.1 主要實驗儀器及試劑
瑞士Bruker DMX-400型核磁共振儀;安捷倫1260II液相色譜儀器;無水乙酸銅(國藥試劑,含量99%);4-二甲氨基吡啶(DMAP)(安徽省郎溪縣聯科實業有限公司,含量99.0%以上);吡啶(國藥試劑,含量98%),其它試劑或溶劑均為市售分析純或化學純。
1.2 合成方法的優化
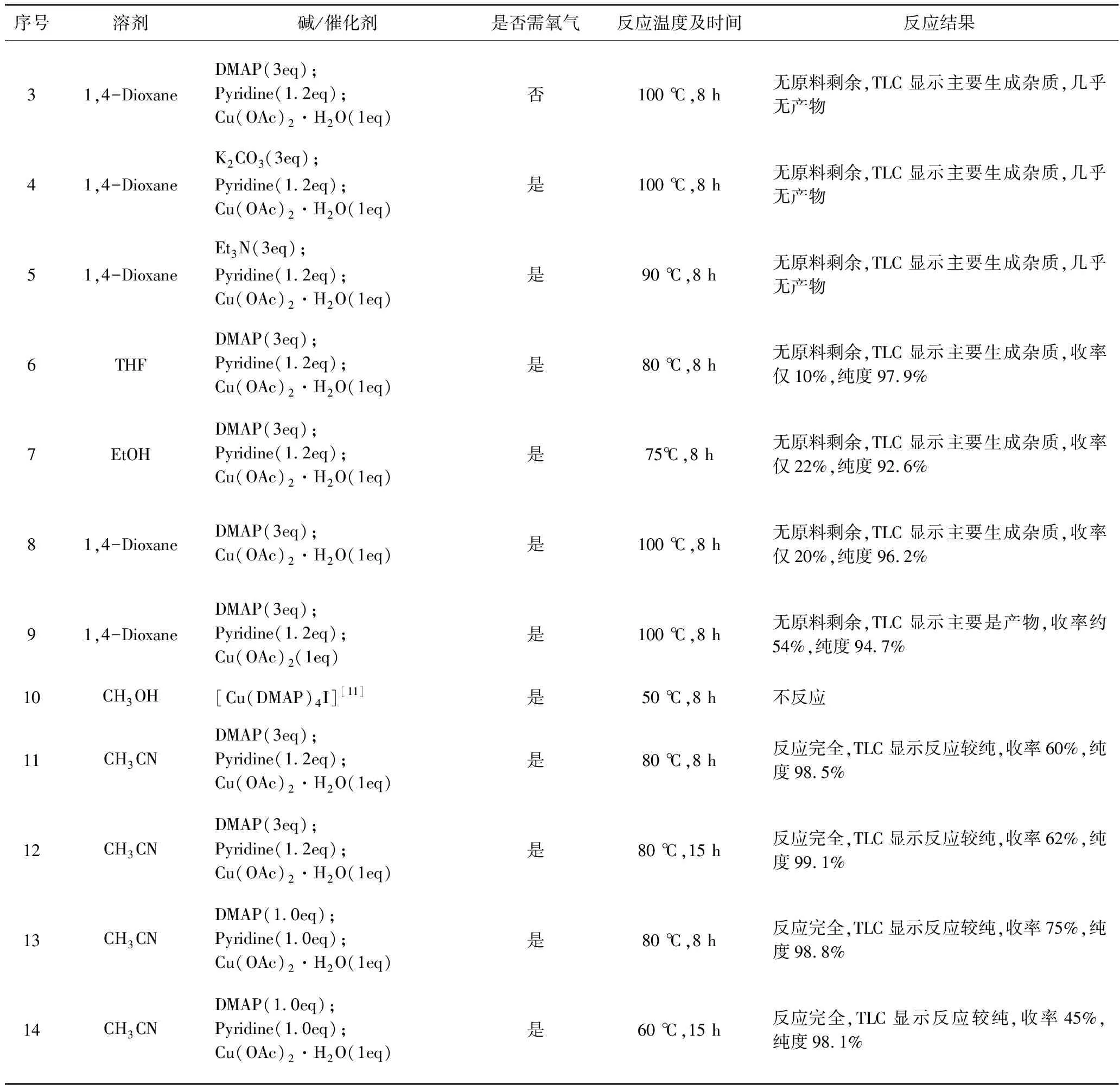
由于目前Chan-Evans-Lam偶聯反應在制備中間體1-環丙基-4-碘-1H吡唑時,均存在著各自的缺點,鑒于此,我們對反應的條件及試劑進行了篩選和優化,結果如表1。

表1 制備1-環丙基-4-碘-1H吡唑反應條件的優化Tab. 1 Optimization of reaction conditions for preparation of 1-cyclopropyl-4-iodo-1H pyrazole
(續表)
2 結果與討論
通過對上述方法的優化,篩選了不同的溶劑,試劑,反應條件,反應時間和反應溫度,結果顯示:
(1) 以DMAP為堿,不通氧氣或反應時間延長(15 h)或反應體系不加吡啶,效果均不好;用無機堿(K2CO3)代替有機堿DMAP,只有少量產物生成;用Et3N代替DMAP,則無產物生成;催化劑用乙酸銅代替一水乙酸銅,效果不明顯。
(2) 參照文獻,以[Cu(DMAP)4I]為催化劑,甲醇為溶劑,則不反應。
(3) 反應溶劑用乙醇,四氫呋喃代替1,4-二氧六環,反應效果均不佳;而用乙腈作溶劑,反應效果較好,在同一溫度下,延長反應時間則收率變化不明顯。
(4) 以乙腈為反應溶劑調整反應試劑的比例,當DMAP,吡啶及一水乙酸銅均為1當量時,反應收率可以達到75%,由此表明堿及催化劑的量應該適中,適當的降低其用量同時調整物料配比反而能增加產物的轉化率。
(5) 當溫度降低至60 ℃時,即使延長反應時間(15 h),最終產物的收率只有45%,由此表明,反應溫度決定了反應的產物轉化率,必須加熱到一定溫度(80 ℃),達到一定的能壘,反應才能更好的進行。
通過上述試驗結果,我們確定了最終的方法,即為:在80 ℃,氧氣氛圍下,以乙腈為溶劑,DMAP和吡啶作堿、乙酸銅為催化劑,其中DMAP、吡啶、乙酸銅的用量均為1當量,反應8 h,最終可以得到純度為98%以上,收率可達75%的1-環丙基-4-碘-1H吡唑。
3 結論
本研究以4-碘-1H吡唑和環丙基硼酸為原料,經改進后的Chan-Evans-Lam 偶聯反應為合成方法,此方法一改常規用的易產生過氧化物的1,4-二氧六環或是劇毒性的1,2-二氯乙烷為溶劑的反應條件,可以完全避免因溶劑原因導致的EHS風險,同時反應溫度和時間適中,工藝可重復高,最終可以得到高純度、高收率的1-環丙基-4-碘-1H吡唑。
