mTOR通路相關的難治性癲癇3例報告及文獻復習
2021-11-11 05:48:32費凌霞李愷煇胡湘蜀李花郭強
臨床神經外科雜志 2021年5期
費凌霞,李愷煇,胡湘蜀,李花,郭強
隨著近年來遺傳學的大力發展,一些藥物難治性癲癇患者在術前評估的同時開始進行基因學的檢測,并發現各式各樣的基因突變。但目前基因診斷并沒有在難治性癲癇術前評估中作為常規檢測,臨床表現為局灶性癲癇而MRI卻呈陰性的患者中有一部分是遺傳相關癲癇。廣東三九腦科醫院收治的3例mTOR通路相關基因突變致難治性癲癇患者,經手術治療后發作得到有效控制,取得了良好效果。本研究對3例患者的臨床資料進行回顧分析,并結合相關文獻復習,探討mTOR通路相關的難治性癲癇的基因突變特點、外科手術治療方案及療效。現報告如下。
1 臨床資料
1.1 病例1 男性,24歲,右利手,9歲起病。發作表現為先兆(右上肢僵硬感),繼而右側肢體強直,發作后期出現拍打、揮舞、蹬踏、翻滾等動作,持續1 min緩解。服抗癲癇藥無效,每月發作數次。家族史:患者的父親15歲出現癲癇發作,30歲以后未見發作;家族中其他成員無癲癇發作史。基因測序:DEPDC5基因雜合變異c.328_329insTA(插入胸腺嘧啶、腺嘌呤),導致氨基酸改變p.G110Vfs*9移碼突變。其父親基因測序檢測到相同的雜合變異(圖1)。術前評估結果:(1)癥狀學,先兆(右肩發僵麻木無力,或咽喉部嗆水感)→右側肌張力障礙→舞蹈徐動(左側肢體及軀干)→發作后咳嗽,發作期腦電圖示無側向性;(2)MRI檢查示左側中央前溝下降支皮層稍厚(圖2);(3)致癇區,邊緣/旁邊緣系統,左側可能性大;(4)SEEG原因,癥狀學中先兆為兩種,其一為右肩部發僵或麻木無力感,需考慮與S1、S2區相關,鑒別運動前區及輔助運動區;其二為咽喉部嗆水感,少見,需考慮植物神經或咽喉部強直,故島葉需要鑒別。癥狀學演變出現肌張力障礙伴對側舞蹈樣動作,需考慮與島葉后部相關,鑒別扣帶回和顳極。給予患者SEEG植入(圖3),發作期起始于C9-11(左側中央前溝下降支后壁),早期擴散至O2-7(左中央蓋部及島前小葉根部)、S11-14(左中央前回腹側部皮層)、Y4-8(左頂蓋)(圖4)。行左額島中央區致癇灶切除術(圖5)。術后病理檢查示:神經元正常排列結構消失,層狀結構紊亂,皮層下白質見散在神經元異位,符合FCD Ⅰ型。術后隨訪患者4年余,均未再出現癲癇發作。

圖1 病例1患者及其父母基因檢查結果
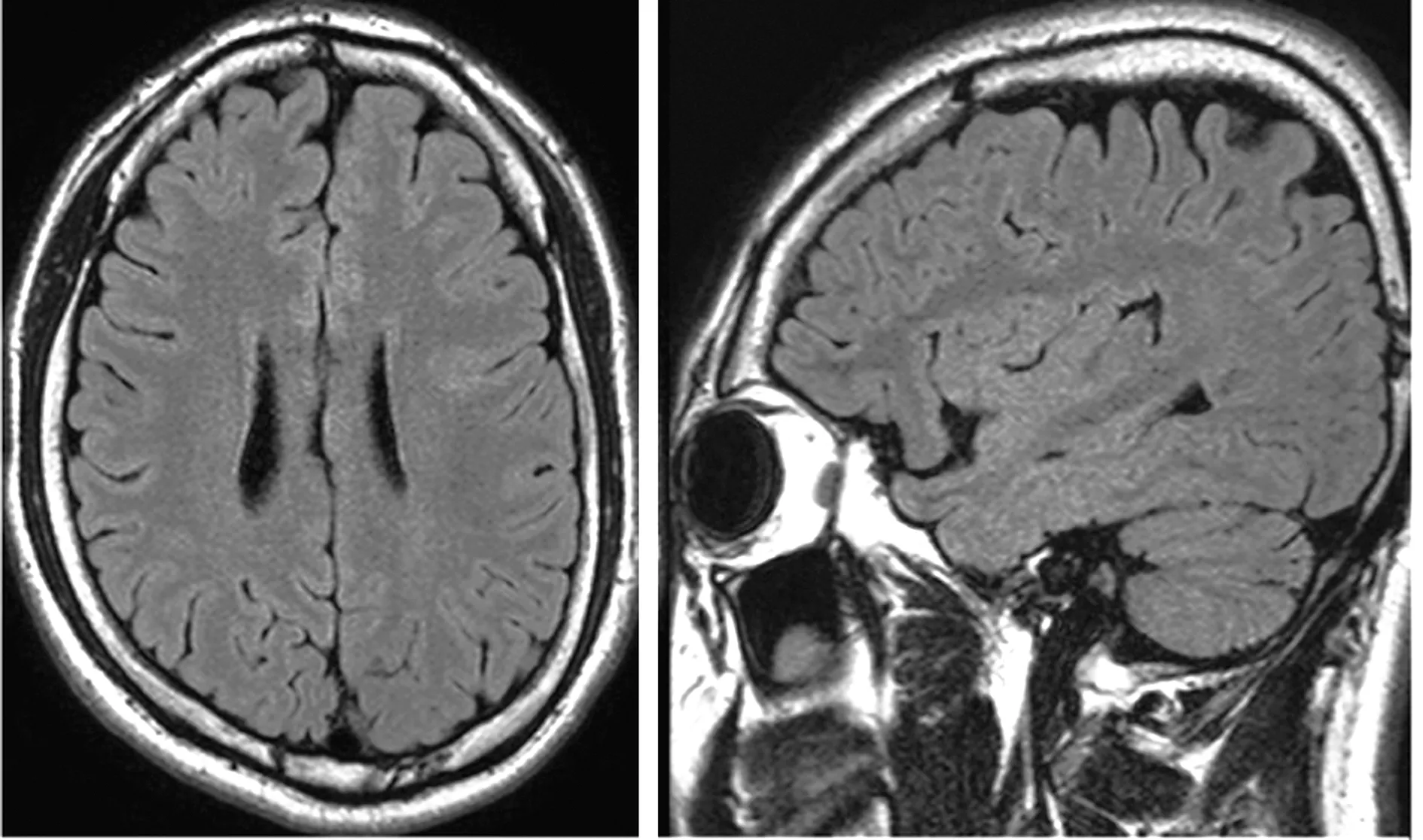
圖2 病例1患者術前頭顱MRI檢查結果

圖3 病例1患者SEEG電極置入示意圖
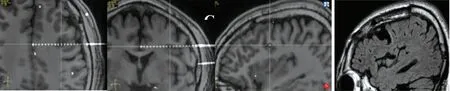
圖4 病例1患者發作起始部位 圖5 病例1患者術后MRI
1.2 病例2 男性,21歲,15歲起病。發作表現:雙耳鳴(低音調、像火車鳴笛)、心慌,隨后出現意識障礙、咂嘴、吞咽、頭眼左側偏轉、口角左歪、左上肢強直,繼之出現全面性強直-陣攣發作;發作2次/月。家族史:患者母親13歲起病,發作表現:雙耳鳴(低音調、像火車鳴笛),隨后出現全面性強直-陣攣發作,發作2~3次/年,未使用抗癲癇藥物治療,30歲后未再出現發作;家族中其他成員均無癲癇發作史。認知心理測試正常范圍。基因測序結果:RELN c.6107C>T(編碼區第6107號核苷酸由胞嘧啶變異為胸腺嘧啶),導致氨基酸改變p.A2036V(第2036號氨基酸由丙氨酸變異為纈氨酸),為錯義突變。其母親基因檢測為相同的雜合突變。術前評估結果:(1)癥狀學,先兆(聽幻覺,心慌)→植物神經癥狀(心率增快)→自動運動(吞咽、雙手)→左手重復運動→雙眼右視→左側偏轉→左側強直-陣攣→全面性強直-陣攣發作,腦電圖提示:發作起始于右側半球(圖6);(2)MRI檢查示右側頂蓋、顳蓋皮層可疑增厚(圖7);(3)致癇區,為右側顳葉聽覺皮層可能性大;(4)SEEG原因,進一步明確致癇灶,并鑒別顳葉及島葉,制定切除邊界。給予患者SEEG植入(圖8),發作起始于Y′4-5(顳橫回),早期擴散至U′1-5(顳橫回及下環島溝后部),W′1-4(顳橫回及下環島溝中部),Y′1-2(島后長回),O′1-2(上環島溝后部),T1-2(下環島溝前部)(圖9)。行右側顳橫回、島后小葉、上下環島溝后部切除術。術后病理診斷為FCD Ⅱa。術后隨訪患者1年,僅出現過1次發作(Engel分級1c)。
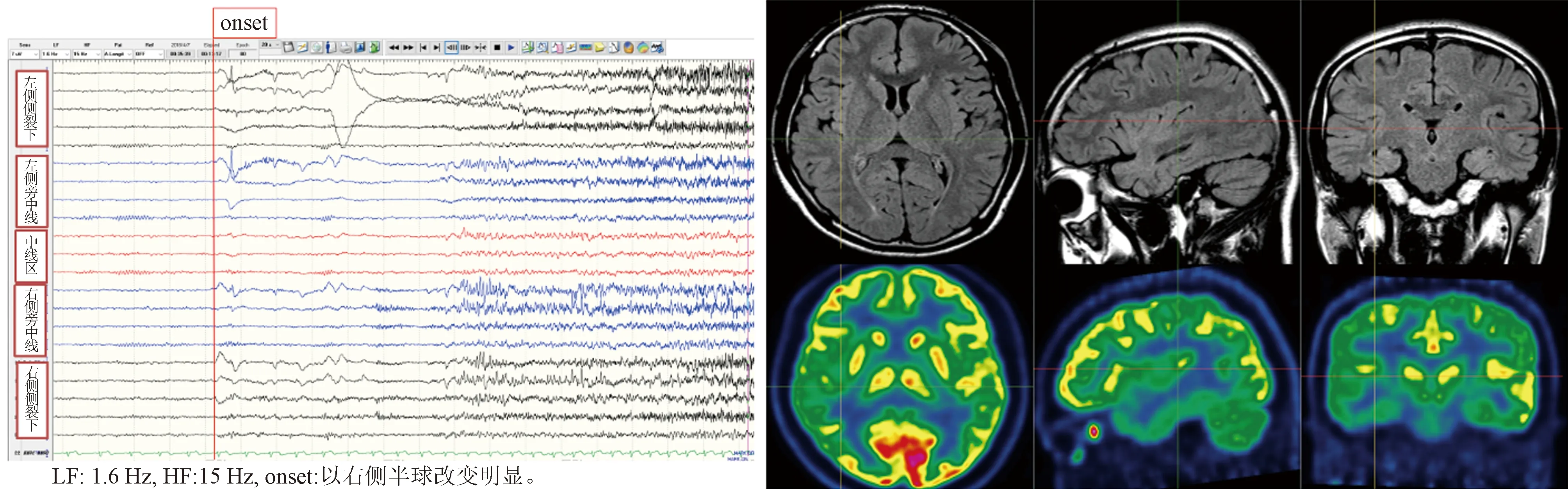
圖6 病例2患者發作起始時腦電圖改變 圖7 病例2患者MRI和PET檢查結果
圖8 病例2患者SEEG電極置入示意圖 圖9 病例2患者SEEG植入后MRI檢查
1.3 病例3 男性,8歲,6歲起病。發作表現:突然發呆、動作停止、頭眼偏向左側、雙上肢展開,伴流涎,發作后期伴咀嚼、吞咽動作,癥狀持續1 min緩解;發作每月8~10次。家族史:家族中無其他成員有癲癇病史,其父親背部皮膚可見數塊色素脫失斑,顱腦MRI檢查未見明顯異常。基因檢測:TCS2基因雜合突變,突變位置:chr16-2130189。其父親基因檢測示相同突變。術前評估結果:(1)癥狀學,動作停止→雙眼左側斜視→復雜運動→強直(軸肌及雙上肢近端)→植物神經癥狀(心率增快)。腦電圖提示:發作起始于左側半球(圖10);(2)MRI,雙側大腦半球多發結節,考慮結節性硬化癥(圖11);(3)致癇區,左側顳葉可能性大;(4)SEEG原因,明確致癇區及網絡,島葉皮層增厚,明確附加癥可能。給予患者SEEG植入(圖12),記錄到2種類型發作。第1種類型:復雜運動→植物神經癥狀→左側凝視→雙側強直;腦電圖起始于T3-9(島后小葉)及V3-8(島前小葉),早期擴散至H1-3、J1-3(海馬)和P1-3(杏仁核)。第2種類型:眨眼→左側凝視→植物神經癥狀→雙側強直;腦電圖起始于F8-12(顳下回后部),早期擴散至J8-12、H7-10(顳中回),P9-12(顳極)及L10-12(舌回外側面)。最后給予左側顳葉及島葉的切除(圖13)。術后病理檢查:符合局灶性皮質發育不良FCD Ⅱb。術后隨訪患者4年余,未出現癲癇發作,正常上學,成績中上等,記憶力較前好轉。
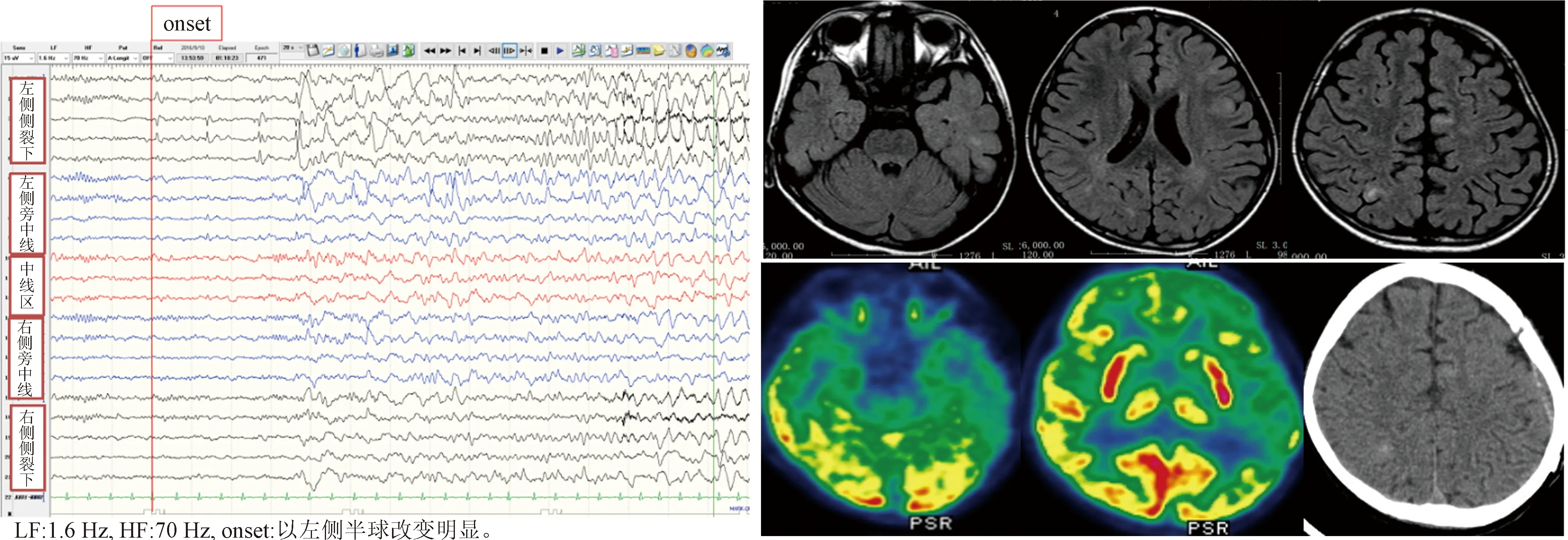
圖10 病例3患者發作起始時腦電圖改變 圖11 病例3患者術前MRI和PET檢查結果

圖12 病例3患者SEEG電極置入計劃
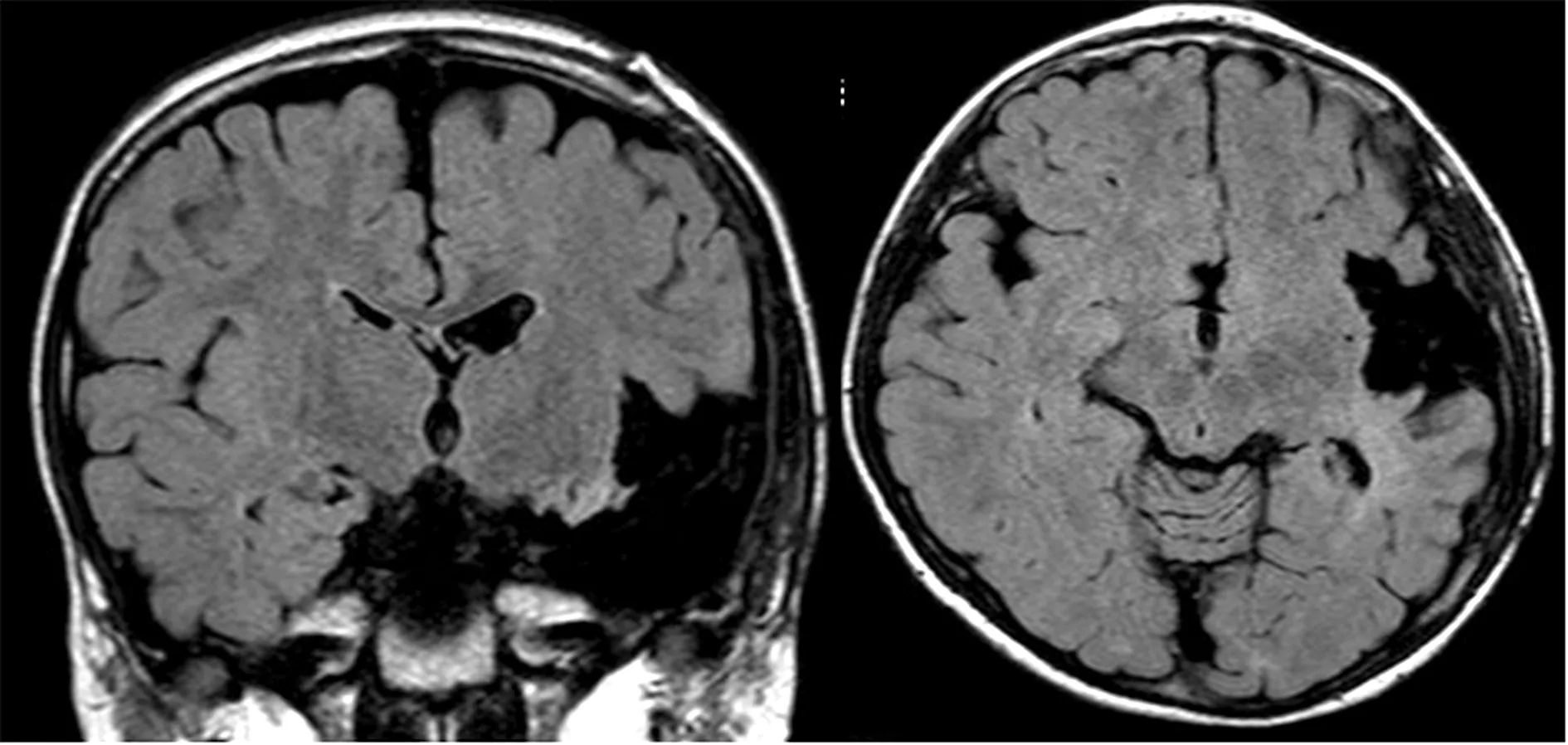
圖13 病例3患者術后MRI檢查結果
2 討 論
2016年廖衛平團隊[1]搜索數個數據庫發現,近1 000種基因突變與癲癇相關;根據基因功能的特點將癲癇相關基因分成離子通道相關基因和非離子通道相關基因,以便更好地認識基因與癲癇的關系,并理解癲癇的發生機制。
DEPDC5基因是目前報道最常見的遺傳性局灶性癲癇的致病基因之一,常染色體顯性遺傳額葉癲癇相關基因中的非離子通道基因。DEPDC5基因定位于染色體22q12,表現為不完全顯性遺傳的特點,主要在神經元中表達[2],致病突變主要表現為截短突變。DEPDC5蛋白是GATOR1復合物的一部分,是mTOR通路中的一個mTORC1負調節劑。Baulac等研究報道59例DEPDC5基因突變患者的MRI表
現,其中皮層發育畸形(malformations of cortical development,MCD)者13例、局灶性皮質不良(focal cortical dysplasia,FCD)9例、溝底型FCD(BOSD)2例、半側巨腦回畸形(hemimegalencephaly,HME)1例、隱匿型皮層下帶狀灰質異位(subcortical band heterotopias,SBH)1例,提示合并FCD甚至HME可能[3],并且出現FCD的部位亦不相同,但以額中央區運動皮質、前額葉內側面皮質較多。DEPDC5基因突變所致癲癇的患者,一般兒童-成人期起病,表型可有差異,大部分智力正常,少數合并神經精神疾病;當發作期癥狀學出現運動系統癥狀時應警惕運動皮質隱匿的FCD。本研究的病例1患者最終證實癲癇起源于中央區。其父親攜帶有相同的基因,而并未出現癲癇發作,也提示種屬變異并不一定會出現癲癇發作;患者同一基因突變卻出現頑固的癲癇發作,且手術后病理檢查示FCD Ⅰ型,表明患者存在二次打擊的體細胞突變;本例患者在手術后病理組織未行更深度的基因測序,無法進一步明確。DEPDC5突變和二次打擊的基因可能引起在錯誤的調控下的體細胞突變的生長,產生皮層的異常。體細胞突變的二次打擊可能是其他DEPDC5的等位基因,或其他mTOR通路上的基因[4]。mTOR通路相關基因變異,此類患者中58%的患者手術后無發作,相對來說效果是比較滿意的[5]。因此,對于外周血檢測到的DEPDC5基因突變者,常提示廣泛的組織受累,所有腦細胞均攜帶突變基因,可以遺傳至下一代;如MRI檢查陰性,基本不建議手術治療,預后不確定;如MRI檢查陽性,提示病灶處有體細胞發生突變(二次打擊),因此可以考慮手術治療。
RELN基因為常染色體顯性遺傳顳葉外側癲癇(autosomal dominant lateral temporal lobe epilepsy,ADLTE),又稱為伴聽覺特征的常染色體顯性遺傳部分性癲癇,青中年起病,發作頻率少,抗癲癇藥物療效好,但撤藥易復發。目前已發現的相關基因主要是LGI-1和RELN基因。LGI-1變異是該病的特異性基因,但僅50%家族呈現LGI-1雜合子變異的典型表型[6],其余部分與RELN基因突變相關。Reelin基因在維持腦皮層的發育中非常關鍵[7],是神經元遷徙過程中的一種停止信號糖蛋白[8]。RELN基因主要可以通過影響神經元移行,導致MCD[9]。有學者在對比RELN基因突變與LGI-1基因突變患者的臨床特點時發現,除更多左側的腦電圖的異常外,其余特點與LG1突變的家族性顳葉癲癇患者未見明顯的區別[10]。本研究的病例2患者有頑固性癲癇發作,致癇灶定位于顳橫回及島葉后部皮層,病理檢查示FCD Ⅱa;患者及其母親的血液檢測發現相同位點的RELN基因突變,組織學基因檢測也顯示相同位點的RELN基因突變,但沒有做更深度的基因測序,沒有發現其他不同位點的突變。RELN是mTOR通路上游的相關基因,對于mTOR通路有重要的調控作用。在mTOR通路中,極低水平的體細胞突變即能引起FCDⅡ型的病理改變,手術成功切除的機會也是非常大的[5]。體細胞突變,也稱嵌合現象,一般發生在生殖細胞以后,通常不遺傳至后代。體細胞突變需要在病變腦組織進行基因檢測方能發現,局灶性MCD中檢測到病理性體細胞突變的機會一般只有41%[11]。基因異常同時在MRI檢查發現相關異常(FCD或MCD等)的患者,往往有更高的手術成功率。
結節性硬化是一種常染色體顯性遺傳病,這種疾病可以累及多個臟器,引起多系統功能異常。超過90%的結節性硬化患者會出現中樞神經系統病變,80%~90%的患者會出現癲癇發作,其中50%的患者出現藥物難治性癲癇[12]。結節性硬化基因異常主要在9號染色體上的TSC1基因或16號染色體上的TSC2基因突變,因編碼不同的蛋白合成從而表現出不同的臨床表型,大約85%的患者可以發現以上基因的突變。該基因是mTOR通路的上游基因。TSC突變可導致相關的皮膚改變,如本研究的病例3患者,其父親攜帶相同的基因,有皮膚改變,顱腦MRI上未見相關結節,智能正常,也沒有出現癲癇發作。已有相關的研究肯定了TSC1、TSC2與FCD或HME突變的聯合,并表明二次打擊的種系和體細胞突變可以導致顱腦結節的出現[11]。外科治療顱內多發的結節性硬化癥患者,術前評估明確責任結節最為重要。雖然結節性硬化患者顱內結節多發,但并不是所有結節均有致癇性;另外,結節可以有致癇性,但結節的周邊組織也可以有致癇性,因而切除范圍的制定非常關鍵,包括MRI上的突出結節(大的、凸出的、鈣化的)在內的腦葉切除較單獨結節切除的療效會更好[13]。若無創檢查有明確的腦區指向性,且相應腦區位于非功能區,該結節位于顱腦凸面,并能在術中進行監測的情況下,手術切除病變往往能取得非常好的療效[14]。
DEPDC5、RELN、TSC基因突變均與mTOR通路相關,且通過二次打擊導致皮層發育的異常,可以考慮外科手術干預。腦結構異常只是表型,并不是病因;各種病因都可以引起腦結構性異常,故影響手術預后的主要因素是病因。病因學是術前評估的首要任務,需要多學科參與診斷分析。在根據解剖-電-臨床定位致癇區之前,首先應進行病因學分析,排除不適合手術的病例。對MRI檢查陰性者,外科手術須謹慎;同時需要進行更加高清的MRI序列檢查,以發現更加微小的腦結構異常[15],提高患者的手術成功率。對于明確的MCD或FCD患者,進行遺傳學檢測仍然有助于確定病因和判斷預后。
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中國民間療法(2021年5期)2021-06-09 09:21:04
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學生導刊(2018年13期)2018-06-29 03:49:00
飲食科學(2017年5期)2017-05-20 17:11:53
現代檢驗醫學雜志(2016年4期)2016-11-15 02:01:14
安徽醫科大學學報(2015年9期)2015-12-16 11:09:44
西南軍醫(2015年4期)2015-01-23 01:19:30
中國中醫藥現代遠程教育(2014年20期)2014-03-01 04:31:21
中國神經精神疾病雜志(2014年1期)2014-03-01 03:23:22
