蜜勒胺催化劑的制備及在環(huán)碳酸酯合成中的應用研究
2021-03-27 09:24:40張源萍李曉云邸亞麗趙雨花亢茂青李其峰王軍威
燃料化學學報 2021年3期
關鍵詞:催化劑
張源萍,李曉云,邸亞麗,趙雨花,亢茂青,李其峰,*,王軍威
(1.中國科學院山西煤炭化學研究所, 山西 太原 030001;2.中國科學院大學, 北京 100049;3.北京機電工程總體設計部, 北京 100000)
當前,二氧化碳 (CO2) 過度排放帶來的溫室效應已引起全球氣候環(huán)境的巨大變化[1],碳減排成為舉世矚目的熱點。在眾多減排方式中,將CO2進行化學固定制備高附加值化學品,是最具經濟效益的一種[2]。由于CO2的高化學穩(wěn)定性,其轉化往往會帶來高能耗或高氫耗,研究者一直在尋找更加經濟可行的轉化途徑,其中,二氧化碳通過環(huán)加成制備環(huán)狀碳酸酯的反應引起大家關注。這一類原子經濟反應的產物環(huán)狀碳酸酯用途廣泛,可應用于溶劑、增塑劑、電解液、聚合物單體、醫(yī)藥等領域[3-5]。目前,已發(fā)現多種催化劑可用于 CO2合成環(huán)狀碳酸酯的反應中,其中,非均相催化劑因易于分離和重復使用而成為研究的重點,已報道的非均相催化劑主要包括金屬氧化物[6, 7]、分子篩[8, 9]、金 屬 有 機 骨 架 (MOF)[10, 11]、 石 墨 相 氮 化 碳 (g-[12, 13]等。
石墨相氮化碳 (g-C3N4) 是一種以三均三嗪環(huán)為基本結構單元的非金屬材料,能夠以尿素、三聚氰胺等含氮有機物為原料進行制備,可應用于CO2的吸附、活化及催化轉化,通過摻入雜原子或改善織構性質的方式可以進一步提高氮化碳的催化活性[14, 15]。三聚氰胺由于價格低廉,且制備氮化碳的收率高,相關的研究報道也很多。在以三聚氰胺為前驅體,熱縮聚合成g-C3N4的過程中(圖1),會相繼產生不同縮聚程度的蜜白胺 (melam)、蜜勒胺 (melem)、大肚胺 (melon) 等中間體,這些中間體均由三嗪結構單元構成,且含有不同類型的胺基,對CO2分子具有較強的活化作用[16, 17],在催化CO2環(huán)加成反應中具有潛在的應用價值。Song等[18]以三聚氰胺為前驅體,在450 ℃熱縮聚得到melem低聚物,并進行羥基和季銨鹽基團接枝改性,低聚物的比表面積和孔體積都有了顯著的增加,對環(huán)氧丙烷與CO2的環(huán)加成反應具有更高的催化活性。在本課題組的前期研究中,以三聚氰胺為前驅體,合成了一系列不同縮聚程度的三嗪單元化合物,隨后將其在水中進行自組裝,得到表面含有?OH的melem催化劑,雖然其比表面積較低 (13.6 m2/g),但在 1,4-丁二醇二縮水甘油醚 (BDODGE)和CO2的環(huán)加成反應中,表現出良好的催化活性,140 ℃,2.0 MPa條件下,BDODGE 轉化率達到93.1%,環(huán)碳酸酯選擇性為99.3%,并表現出優(yōu)異的循環(huán)使用穩(wěn)定性[19]。

圖1 類石墨相氮化碳 (g-C3N4) 的合成過程Figure 1 Synthesis reaction process of g-C3N4
為了進一步認識melem催化劑的構效關系,探索催化劑活性提高的途徑,本研究采用納米二氧化硅為模板劑進行melem催化劑的制備,以NaOH溶液脫除模板劑,改善催化劑的織構性質和表面性質,提高比表面積和表面活性位數量。將melem作為催化劑用于1,4-丁二醇二縮水甘油醚(BDODGE) 與 CO2的環(huán)加成反應,制備可用于聚氨酯、聚碳酸酯等高分子聚合物的多官能度環(huán)狀碳酸酯。系統(tǒng)考察了melem材料在反應過程中的催化性能,并結合XRD、FT-IR、N2吸附-脫附等表征技術研究了模板劑對melem催化劑結構和性能的影響。
1 實驗部分
1.1 原料與試劑
主要試劑:三聚氰胺(99%,阿拉丁試劑(上海)有限公司);納米二氧化硅(99.5%,50±5 nm,麥克林);濃鹽酸、丙酮、甲苯(分析純,國藥集團化學試劑有限公司);氫氧化鈉(分析純,天津市風船化學試劑科技有限公司);1,4-丁二醇二縮水甘油醚(BDODGE)(環(huán)氧值:0.75 mol/100 g)(武漢遠成共創(chuàng)科技有限公司);二氧化碳(99%,山西宜虹氣體工業(yè)有限公司)。
1.2 催化劑的制備
1.2.1 melem催化劑的制備
稱取一定量的三聚氰胺于帶蓋的坩堝中,置于馬弗爐中升溫至450 ℃ 焙燒4 h(升溫速率5 ℃/min),得到淡黃色固體melem產物,研磨成細粉后留存待用,標記為CN-450。
1.2.2 介孔melem催化劑的制備
將準確稱量的納米二氧化硅加入三聚氰胺粉體中研磨混合均勻,之后將其放入帶蓋的坩堝中,置于馬弗爐中升溫至450 ℃ 焙燒4 h(升溫速率5 ℃/min),降至室溫后取出,所得樣品標記為CN-450-r(r表示三聚氰胺與納米SiO2球的質量比)。之后,將一部分樣品粉末分散于 100 mL 的 2 mol/L NaOH 溶液中,室溫攪拌8 h,除去SiO2模板劑。用去離子水將其洗滌至中性,再用無水乙醇洗滌三次,最后110 ℃烘干,得到介孔melem材料,標記為mp-CN-450-r(r表示三聚氰胺與納米SiO2球的質量比)。本次實驗制得的產物有:CN-450、CN-450-2、CN-450-4、CN-450-8,以及對產物除模板后得到的相應產物為:mp-CN-450-2、mp-CN-450-4、mp-CN-450-8。其合成過程示意圖見圖2。

圖2 蜜勒胺 (melem) 催化劑的合成路線示意圖Figure 2 Synthesis process of melem catalyst
1.3 催化劑的評價
以 1,4-丁二醇二縮水甘油醚 (BDODGE) 與CO2的環(huán)加成反應為活性評價反應(如圖3),考察催化劑的催化性能。具體操作過程如下:將30 g BDODGE 和 1.5 g催化劑加入到 100 mL 不銹鋼高壓釜中,向反應釜中緩慢通入N2氣體置換釜內空氣。之后密閉反應釜,將反應釜程序升溫至反應溫度后,通入CO2,保持在2.0 MPa壓力下進行反應。反應20 h后,停止反應,冷卻至室溫后緩慢排出未反應的CO2。催化劑和反應產物采用過濾及離心方法進行分離,催化劑回收待用,收集濾液進行分析。采用FT-IR和1H NMR對產物結構進行表征,采用鹽酸-丙酮化學滴定及1H NMR表征進行轉化率和環(huán)狀碳酸酯選擇性的定量分析,具體方法參見文獻[20]。

圖3 BDODGE 與 CO2 的環(huán)加成反應Figure 3 Cycloaddition reaction of BDODGE with CO2
1.4 催化劑的表征
采用 X 射線粉末衍射儀 (XRD,D8 advance,德國 Bruker 公司)表征催化劑的晶體結構,CuKα 光源輻射,2θ為 5°?80°,掃描步長為 0.02°,掃描速率為 2(°)/min;采用傅里葉變換紅外光譜儀(FT-IR,Nicolet 380,美國 Thermo electron 公司)進行結構分析,催化劑以KBr壓片法來制樣分析,液體產物采用KBr片涂膜法進行分析,儀器的分辨率和波譜測試范圍分別為 4.0 cm?1及 4000?400 cm?1;采用掃描電子顯微鏡(SEM,JSM-7001F,日本電子公司)對樣品的表面形貌進行觀察,加速電壓為5 kV;采用 TriStar II (3020)物理吸附儀 (美國 Micromeritics公司)來表征催化劑的織構特征,吸附質為N2,吸附溫度?196 °C,采用 Brunauer-Emmett-Teller (BET)方程式計算樣品比表面積;采用氫核磁共振儀 (1H NMR,AVANE 400 MHz,瑞士 Bruker)對反應物及產物成分進行定性和定量分析,選用氘代氯仿作為溶劑,甲苯作為內標物。
2 結果與討論
2.1 催化劑的結構表征
2.1.1 XRD表征
圖4(a)為不同三聚氰胺與二氧化硅質量比條件下制備得到的含有二氧化硅樣品的(CN-450-r)的XRD譜圖。為方便分析,將無模板條件下制備的樣品 (CN-450) 的XRD結果也列于圖4中。CN-450樣品的譜圖中,在 12.5°、13.6°、16.7°、18.4°、19.7°、22.1°、25.3°、27.3°和 30.5°等處出現衍射峰,這些衍射峰與文獻報道的melem物相[21]的衍射峰一致。與CN-450相比,引入模板劑納米SiO2的CN-450-r樣品,其melem衍射峰強度明顯變弱,而且隨著模板劑質量的增加,其衍射峰的強度逐漸減弱,CN-450-2樣品僅有微弱的衍射峰。這一方面是由于SiO2的摻入使得產物中melem組分的含量減少;另一方面,這種納米SiO2顆粒細小且具有多孔結構,能夠促進melem的分散,并阻止了melem顆粒間的聚集和晶體的生長。由圖4(b)可知,脫除模板后樣品 (mp-CN-450-r) 的晶體結構與脫除前樣品 (CN-450-r) 有較大變化,在 6.1°與 12.2°處出現了新的衍射峰,分別對應于melem水合相的 (110) 和(220) 晶面[22]。這種水合相是在NaOH溶液去除模板劑的過程中形成的,與melem在水中的自組裝行為有關。此外,與去除模板前樣品相比較,去除模板后樣品的melem相衍射峰的位置基本保持不變,這表明mp-CN-450-r樣品也是由melem物相組成,并可能在melem樣品表面產生部分羥基。
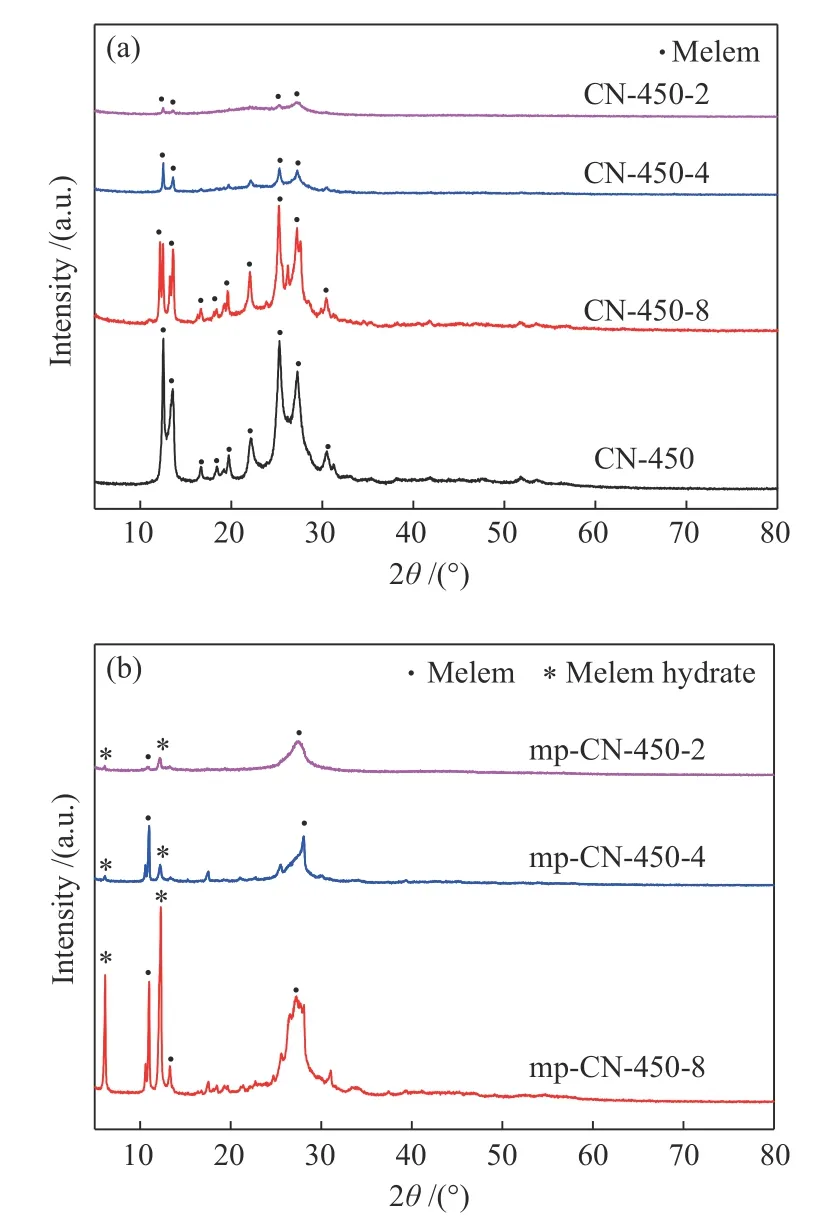
圖4 樣品 CN-450-r (a) 和 mp-CN-450-r (b) 的 XRD 譜圖Figure 4 XRD patterns of CN-450-r (a)and mp-CN-450-r (b) catalysts
2.1.2 FT-IR表征
為了進一步確定CN-450-r及mp-CN-450-r催化劑的組成和表面結構,對樣品進行了紅外表征,結果見圖 5。在 CN-450樣品的譜圖中,1617、1458以及807 cm?1處出現較強的紅外吸收峰,可歸屬于三嗪環(huán)結構中C?N或C=N的伸縮振動和彎曲振動。在3470和3418 cm?1處較弱的紅外吸收,對應于胺基N?H的伸縮振動。這些表明,CN-450樣品中存在melem物相。與CN-450相比,CN-450-r在1099 cm?1處的紅外吸收峰的強度明顯增強,這是由于模板劑SiO2的Si?O鍵的不對稱伸縮振動峰和歸屬于melem結構中伯胺與環(huán)相連的C?N的伸縮振動峰重疊所導致的,此外,在466 cm?1處出現新的紅外吸收峰可對應于Si?O鍵的對稱伸縮振動,表明CN-450-r中有SiO2的摻入。并且隨著SiO2摻入量的增加,這兩個吸收峰也逐漸增強。去除模板樣品的FT-IR譜圖5(b)與去模板前樣品CN-450-r相比,去模板后mp-CN-450-r的譜圖中位于 1099 和 466 cm?1兩處 Si?O 振動吸收峰消失,表明模板劑去除完全。除去模板劑后,所有樣品在 1617、1458 以及 807 cm?1處也出現了較強的歸屬于melem結構中C?N的紅外吸收峰,這表明mp-CN-450-r樣品中不含SiO2,結合XRD結果可以確定,mp-CN-450-r樣品主要是由melem物相組成的。
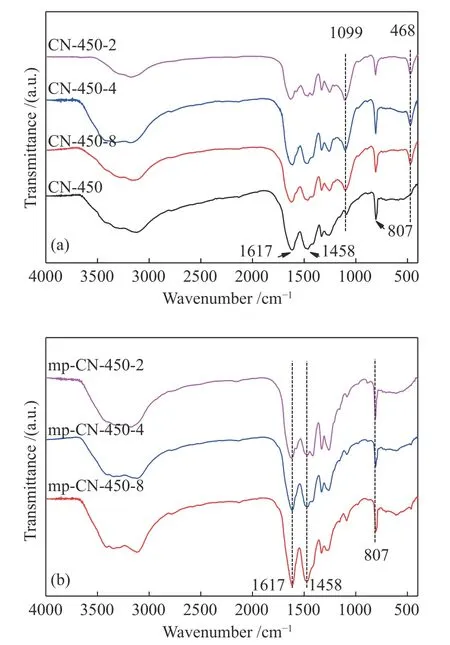
圖5 樣品 CN-450-r (a) 和 mp-CN-450-r (b) 的 FT-IR 譜圖Figure 5 FT-IR spectra of CN-450-r (a)and mp-CN-450-r (b) catalysts
2.1.3 表面形貌表征
為了研究催化劑的表面形貌,對所制備的催化劑進行了掃描電鏡的測試。圖6(a)為CN-450的SEM照片,可以觀察到樣品CN-450無固定的形貌,其催化劑粒子為緊密堆積的塊狀結構。與CN-450不同,mp-CN-450-r樣品表面為海綿狀的多孔疏松結構,mp-CN-450-r的特殊結構是由模板劑納米SiO2引起的。在使用NaOH將模板劑脫除后,樣品中被SiO2占據的位置形成新的孔道,從而顯示為多孔結構。

圖6 CN-450 (a) 和 mp-CN-450-r (b) 樣品的 SEM 照片Figure 6 SEM images of CN-450 (a)and mp-CN-450-r (b) samples
2.1.4 BET表征
圖7為mp-CN-450-r催化劑的N2吸附-脫附等溫線及對應的孔徑分布曲線。由圖7可知,樣品mp-CN-450-r的氮氣吸附-脫附等溫線是典型的IV型等溫曲線,表明該催化劑具有典型的介孔結構。運用BJH模型計算催化劑的孔徑可知,mp-CN-450-r的孔徑主要分布在28 nm左右,然而所采用的模板劑納米 SiO2的尺寸為(50±5 )nm,這種變化一方面是去除模板劑過程中melem發(fā)生自組裝引起的;另一方面mp-CN-450-r在焙燒的過程中也形成了一種具有裂孔的堆疊片層結構[23],這些都使得產物的孔道結構發(fā)生改變,同時比表面積顯著增大。
表1為系列CN-450-r及mp-CN-450-r樣品的N2吸附-脫附測試結果。對比CN-450和CN-450-r可知,模板劑納米SiO2的摻入顯著提高了樣品的比表面積和孔體積,這是由于納米SiO2本身就是一種多孔材料,在焙燒過程中,部分三聚氰胺發(fā)生氣化,進入SiO2孔道內發(fā)生氣相沉積,堵塞SiO2孔道;大部分會在SiO2顆粒間聚合、重組,構成復雜的孔道結構。對于CN-450-r樣品而言,隨著SiO2摻入量的增加,樣品的比表面積和孔體積逐漸增大,但由于SiO2孔道被堵塞,比表面積明顯低于SiO2。脫除模板劑后,SiO2占據的位置形成新的孔道,樣品 (mp-CN-450-r) 的比表面積和孔體積都有顯著提高。mp-CN-450-2的比表面積可達92 m2/g,比CN-450提高20多倍,也比前期所制備的催化劑CN-450-W提高近七倍[19]。更大的比表面積無疑會增大催化劑與反應物的接觸、增加反應活性位點,有利于提高催化活性。
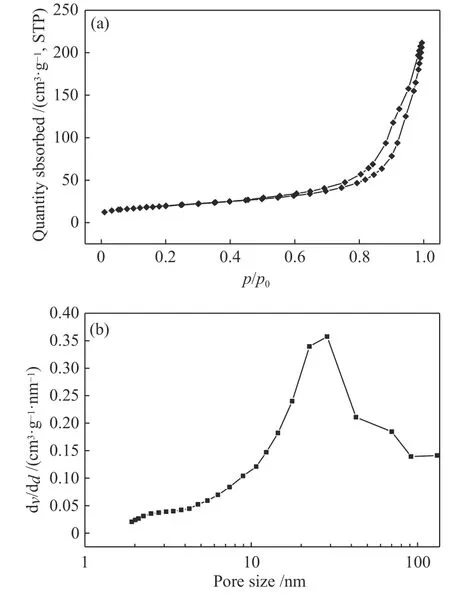
圖7 mp-CN-450-r催化劑 N2 吸附-脫附等溫線 (a)和孔徑分布 (b)Figure 7 Nitrogen adsorption-desorption isotherms (a) and Barret-Joyner-Halenda (BJH) pore size distribution plots (b)of mp-CN-450-r
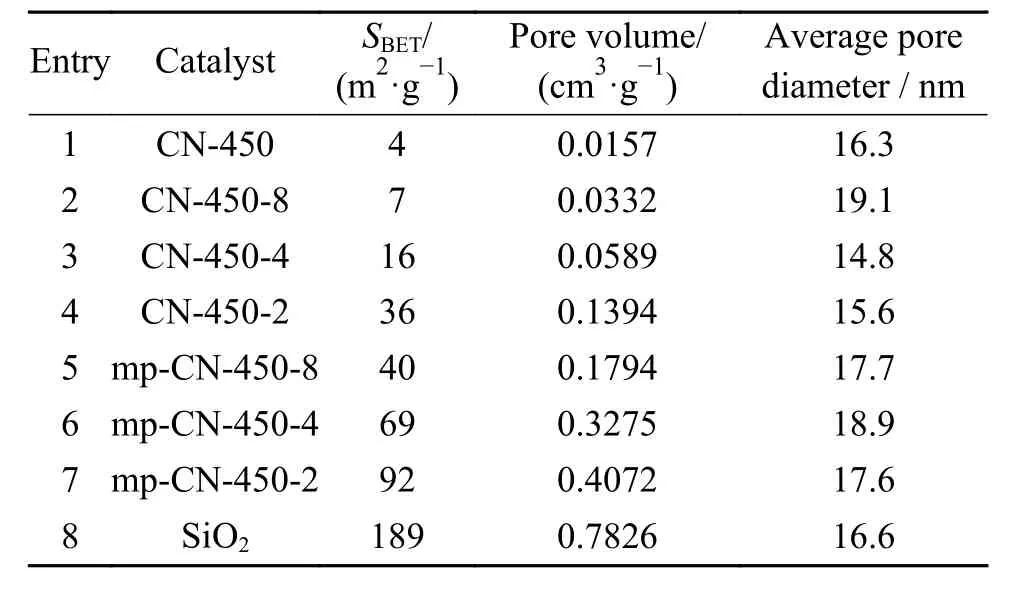
表1 CN-450-r及mp-CN-450-r材料的織構分析Table 1 Textual parameters of CN-450-r and mp-CN-450-r materials
2.2 產物結構表征
圖8為1,4-丁二醇二縮水甘油醚 (BDODGE)及其環(huán)狀碳酸酯產物 (Cyclic Carbonate)的結構分析結果。圖8(a)為兩者的FT-IR譜圖,如圖8(a)所示,2925 和 2870 cm?1處出現的紅外吸收峰分別歸屬于結構中的亞甲基及甲基的C?H伸縮振動;1107 cm?1處的強峰對應的是 C?O?C 的伸縮振動。BDODGE 與 CO2反應之后,在 915 cm?1處歸屬于環(huán)氧基團的吸收峰消失,相應的在1798及1174 cm?1出現新的吸收峰,分別歸屬于環(huán)狀碳酸酯基團中的C=O和C?O的振動峰。圖8(b)為BDODGE及其環(huán)加成產物五元環(huán)狀碳酸酯的1H NMR表征譜圖。在環(huán)狀碳酸酯的譜圖中,環(huán)氧基團在 2.61?3.15 (δa= 2.61、δb= 2.80、δc= 3.15) 處的化學位移峰完全消失,而在 4.39?4.82 (δa'= 4.39、δb'=4.50、δc'= 4.82) 出現新的位移峰,這些新的位移峰歸屬于生成的環(huán)狀碳酸酯基團的亞甲基和次甲基質子峰。以上結果均證明1,4-丁二醇二縮水甘油醚與CO2反應后完全轉化為1,4-丁二醇二縮水甘油醚型五元環(huán)狀碳酸酯。
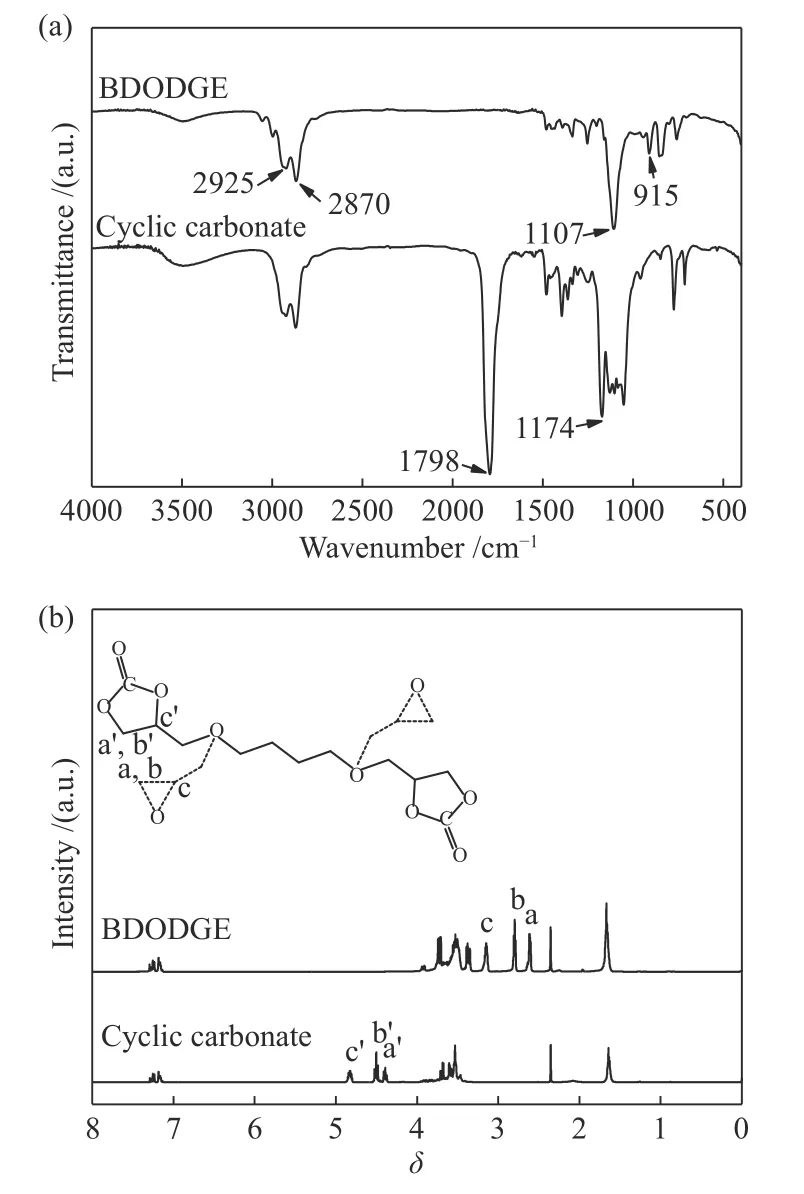
圖8 BDODGE 及相應的環(huán)狀碳酸酯的 FT-IR 和1H NMR 譜圖Figure 8 FT-IR spectra and 1H NMR spectra of BDODGE and cyclic carbonate
2.3 催化劑活性評價
將不同質量比制得的催化劑應用于1,4-丁二醇二縮水甘油醚 (BDODGE) 和CO2的環(huán)加成反應中,反應結果見表2。
由表2可知,無模板條件下制備的樣品 (CN-450),催化活性較低,其原因可能是催化劑的比表面積較小,活性基團與反應物分子未能充分接觸所導致的。對于去除模板前的樣品(CN-450-r) 而言,其催化活性僅稍有提高,主要是由于模板劑SiO2摻入雖然提高了樣品的比表面積,但也覆蓋了melem的活性中心,減少了活性中心與反應物的接觸。而去除模板劑后的樣品 (mp-CN-450-r),催化活性均顯著提高,這主要有兩方面的原因:一方面是模板劑的去除使得催化劑擁有較豐富的孔結構,獲得較高的比表面積,使得催化劑可以和反應物分子更好的接觸,有利于反應的進行;另一方面,催化劑中的melem水合相的產生使得反應過程中有?OH的參與,據文獻報道,?OH可以與環(huán)氧化合物的氧原子形成氫鍵,從而極化環(huán)氧化合物的C?O鍵,促使環(huán)氧化合物活化開環(huán),從而促進環(huán)加成反應的進行[24]。與前期催化劑CN-450-W[19]相比,mp-CN-450-2的催化活性更高,在用量為 5% 時,130 ℃ 反應 20 h,BDODGE 的轉化率可達99.3%。這表明,催化劑比表面積的提高,使得BDODGE可以在較低的反應溫度下完成環(huán)加成反應。此外,催化劑表面形成的羥基也對環(huán)氧基團的活化和環(huán)狀碳酸酯的生成具有一定的促進作用,它與含N堿中心的協同催化作用,共同提高了melem催化劑的活性。
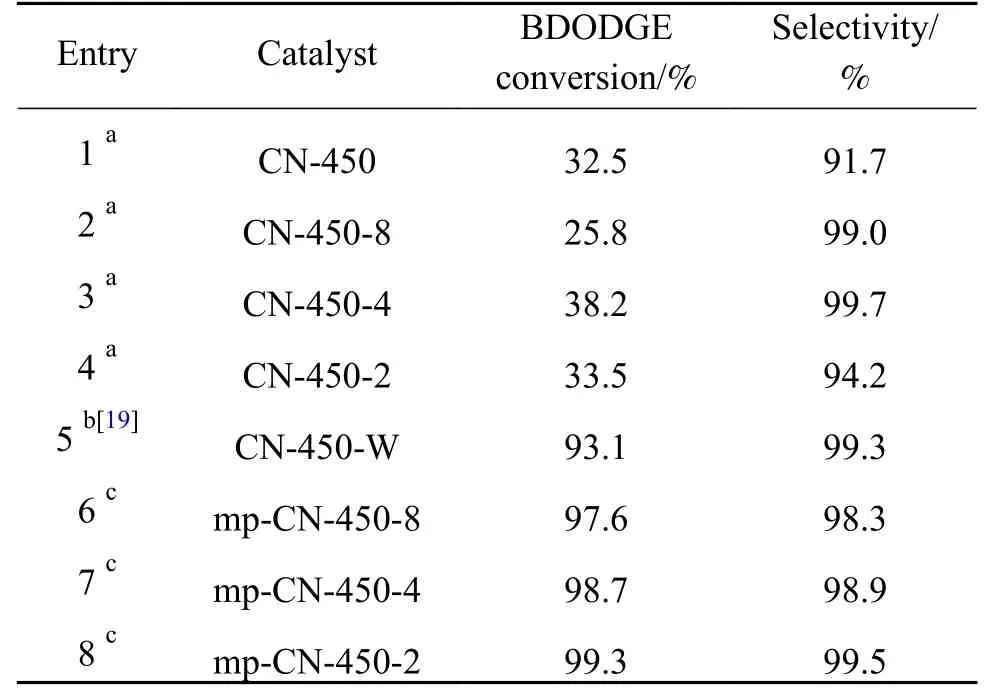
表2 CN-450-r及mp-CN-450-r催化劑對BDODGE與CO2環(huán)加成反應的影響Table 2 Catalytic performance of different CN-450-r and mp-CN-450-r catalysts in the reaction of BDODGE and CO2
2.4 不同催化劑催化CO2化學轉化的活性對比
將mp-CN-450-2的催化性能與已報道的催化劑進行了對比,結果見表3。以三聚氰胺為原料制備的m-C3N4作為催化劑時,BDODGE僅得到36.1%的產率,其原因可能是塊狀催化劑比表面積 (3 m2/g)較小,活性基團與反應物分子不能充分接觸;以尿素為原料制備的u-C3N4催化劑,由于制備過程中產生的大量CO2氣體可作為軟模板劑,使得u-C3N4擁有較大的比表面積 (37 m2/g),從而提高催化劑的催化活性;催化劑ZnBr2/u-CN/r-Al2O3具有高效的催化性能,可達到98.9%的BDODGE產率,但該反應需要在較長的時間 (30 h) 和較大的催化劑用量 (15.1 %) 下進行;離子型交換樹脂 D296 為催化劑,催化聚丙二醇二縮水甘油醚 (PPGDGE) 與CO2環(huán)加成反應時,可得到95.8%的PPGDGE產率,但該反應也需要較長的反應時間,并且反應前需要對D296進行24 h的熱處理;以均相LiBr作為催化劑時,在溫和的反應條件下雙酚A型環(huán)氧樹脂E51的轉化率為94.0%,表現出較高的催化活性,但是,均相催化劑LiBr的分離過程比較繁瑣,這限制了該催化劑的使用。與上面所列的催化劑相比,mp-CN-450-2可以在相對溫和的條件下催化該反應,并得到較高的產率。因此,與已有的催化劑相比,mp-CN-450-2表現出更好的催化性能。
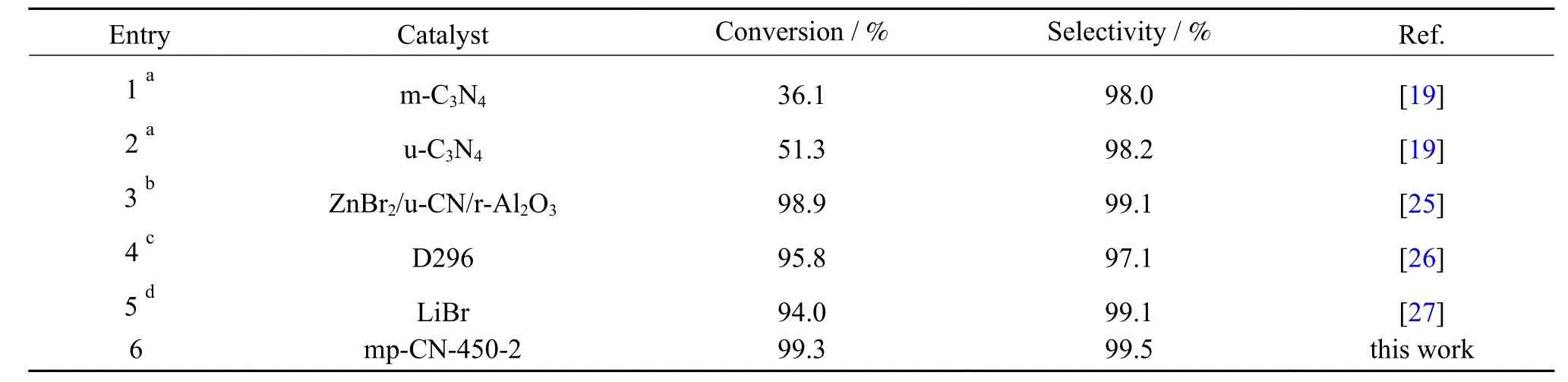
表3 各種催化劑催化CO2環(huán)加成反應對比Table 3 Comparison of various catalysts for the cycloaddition of CO2
3 結 論
本研究采用硬模板劑法制備了系列不同比表面積和孔體積的介孔melem材料,將其應用于環(huán)氧化合物與CO2環(huán)加成合成環(huán)狀碳酸酯的催化轉化反應中,考察了模板劑的引入對melem材料結構性質和催化性能的影響。研究結果表明,采用模板劑可以顯著改善melem材料的織構性質,提高材料的比表面積和孔體積,比表面積可達92 m2/g,是未采用模板劑樣品的23倍;模板劑法不僅改善了催化劑的織構性質,并引入活性羥基,有效提高了催化活性,在 130 ℃、2.0 MPa 條件下,反應 20 h后,BDODGE的轉化率可達99.3%,環(huán)狀碳酸酯的選擇性為99.5%。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
