PRRT2 c.649C>T突變導致智力障礙以及蛋白質表達異常 *
2021-01-29 02:23:52馮浩洋胡平喬鳳昌王艷許爭峰
臨床檢驗雜志 2020年12期
關鍵詞:癲癇
馮浩洋,胡平,喬鳳昌,王艷,許爭峰
(南京醫(yī)科大學附屬婦產醫(yī)院&南京市婦幼保健院產前診斷中心,南京210004)
自從2011年在人類中發(fā)現(xiàn)富含脯氨酸的跨膜蛋白2 基因(PRRT2)(染色體16p11.2)的第1個突變以來,該基因突變被認為與多種神經系統(tǒng)疾病有關,如陣發(fā)性運動性運動障礙(OMIM#128200)、良性家族性嬰兒癲癇(OMIM #605751)等[1]。此外,由于該基因突變與運動系統(tǒng)癥狀密切相關,但智力障礙癥狀往往難以被聯(lián)系在一起,運動障礙同時伴有智力障礙的病例報道極為罕見,因此可能導致一些存在智力發(fā)育缺陷但運動系統(tǒng)癥狀輕微的PRRT2突變患者漏診。近年來,外顯子測序技術的推廣使診斷單基因變異所致的遺傳病更為便捷、準確[2]。本研究利用外顯子測序技術診斷1例智力發(fā)育障礙患者,該患者PRRT2基因存在c.649C>T雜合無義突變,并且在體外實驗證實該突變會導致編碼的PRRT2蛋白不能正常表達。報道如下。
1 材料與方法
1.1研究對象 患者,漢族,男性,14歲,江蘇鹽城人。于2017年7月因認知障礙就診于南京醫(yī)科大學附屬婦產醫(yī)院遺傳中心,進行遺傳咨詢和進一步評估。患者為足月順產,出生5個月后首次病發(fā)癲癇,程度較輕。2004年2月于鹽城市中醫(yī)院檢查腦CT和腦電圖,均提示正常,后服用中藥調理。2006年6月于江蘇省人民醫(yī)院診斷為自閉癥。后患者偶有癲癇發(fā)作,一般由突然站立引起,常僅持續(xù)數秒,并伴有輕微運動障礙。于我院就診時,患者面色呆滯,站立姿勢不穩(wěn),雙手彎曲不自然,反應遲緩,有輕微言語溝通障礙,情緒管理不良。據父母描述,該患兒自理能力較差,缺乏創(chuàng)造性想象力。智商檢測(IQ)為70。本研究通過南京醫(yī)科大學附屬婦產醫(yī)院醫(yī)學倫理委員會審核批準(No.寧婦倫字[2017] KY-081號),所有研究對象均簽署知情同意書。
1.2細胞系、主要儀器及試劑 HEK 293T細胞購自ATCC細胞庫。S220型Covaris超聲波破碎機(美國Covaris公司),Veriti梯度PCR儀(美國ABI公司),Qubit3.0熒光儀(美國Thermo公司),Agilent 2100生物分析儀(美國Agilent公司),Hiseq2500測序儀(美國Illumina公司),Tanon 5200化學發(fā)光成像儀(上海天能公司)。DNA提取試劑盒(天根生物公司),文庫構建試劑盒(上海德儀東方公司),Agilent SureSelect QXT All Human Exon V6試劑盒(美國Agilent公司),PCR擴增試劑盒(南京諾唯贊公司),PrimeSTAR?HS DNA聚合酶(日本TaKaRa公司),無連接克隆試劑盒(南京ABM公司),DMEM培養(yǎng)基(美國Gibco公司),RIPA裂解液(上海碧云天公司),Loading緩沖液(上海天能公司), 蛋白酶抑制劑(瑞士Roche公司),兔抗鼠Prrt2多克隆抗體(美國Sigma公司),兔抗鼠FLAG多克隆抗體(美國CST公司);羊抗兔IgG(H+L)-HRP二抗(南京巴傲得公司)。
1.3外顯子測序 分別抽取患者及父母入院時的外周血5 mL,使用EDTA-K2抗凝。提取先證者及父母外周血中基因組DNA并進行遺傳分析,所有DNA濃度>50 ng/μL,總量>1.5 μg。通過超聲波從剪切的樣品中創(chuàng)建片段文庫,并按照Agilent SureSelect QXT All Human Exon V6試劑盒說明書進行靶向富集。捕獲的DNA被擴增,行固相橋擴增,并在HiSeq2500測序儀上進行雙端測序。使用基因組分析工具包3.4(GATK, www.broad institute.org/gatk)進行與對人類參考序列(hg19)的讀取和變體檢測比對。最后用PCR和Sanger測序驗證突變。針對點突變所在的Exon2的擴增引物由Primer-BLAST在線軟件設計,由蘇州金唯智公司合成(正向引物:5′-TGCCGCTGTCTCTGCTATTC-3′和反向引物:5′-TAAGCGAAGGCCACGATGTT-3′)。PCR產物送蘇州金唯智公司進行一代測序驗證。
1.4構建質粒 為更容易構建質粒,選用小鼠的基因序列。從本課題組前期研究的成年小鼠腦中提取總RNA,反轉錄出第一鏈cDNA[3]。以cDNA為模板使用PrimeSTAR擴增全長小鼠Prrt2編碼序列(正向引物:5′-ATGGCAGCCAGCAGCTCT-3′和反向引物:5′-TTACTTATACACGCCTAA-3′,設計及合成公司同1.3),通過重疊PCR在Prrt2的蛋白質羧基端插入1個FLAG表位以便于后續(xù)的蛋白質檢測,利用無連接克隆試劑盒克隆至pCAGGS-GFP載體構建Prrt2-FLAG質粒。使用野生型Prrt2編碼序列進行定點誘變重疊PCR以獲得Prrt2c.667C>T(p.R223X)序列[對應人PRRT2c.649C>T(p.R217X)](前段序列正向引物:5′-ATGGCAGCCAGCAGCTCT-3′,反向引物:5′-CTGCTGCAGCAGCACTCGGGG-3′;后段序列正向引物:5′-CCCCGAGTGCTGCAGCAG-3′,反向引物:5′-TTACTTATACACGCCTAA-3′,設計及合成同上),利用無連接克隆試劑盒克隆至pCAGGS-GFP載體構建PRRT2mut-FLAG質粒。通過測序整個編碼區(qū)域確認構建的突變質粒。
1.5western blot HEK 293T細胞用含10%胎牛血清的DMEM培養(yǎng)基,置于37 ℃、5%CO2條件下培養(yǎng),使用Lipofectamine 2000試劑按說明書分別瞬時轉染野生型及突變Prrt2質粒,轉染6 h后更換為原培養(yǎng)基繼續(xù)培養(yǎng)。48 h后,細胞在混合蛋白酶抑制劑(1∶100稀釋)的RIPA裂解液中裂解。4 ℃溫育1 h后,將裂解物4 ℃、13 800×g離心10 min。取野生型Prrt2上清液作為對照組,相同劑量和濃度的突變型Prrt2上清液作為實驗組,將上清液與Loading緩沖液(1∶5稀釋)混合煮沸。分別將混合物加載至10%十二烷基硫酸鈉聚丙烯酰胺凝膠上電泳(90 V、20 min,100 V 1 h)。隨后將蛋白質條帶在100 V、200 mA的條件下電轉移至0.22 μm聚乙二醇氟化膜中2 h。室溫條件下置于TBST溶解的50 g/L脫脂奶粉溶液封閉1 h。裁膜后分別加入兔抗鼠Prrt2多克隆抗體(1∶2 000稀釋)以及兔抗鼠FLAG多克隆抗體(1∶1 000稀釋)4 ℃溫育過夜。蛋白質條帶使用TBST洗膜1 h,再加入羊抗兔IgG(H+L)-HRP二抗(1∶20 000稀釋)溫育1 h。在曝光前使用ECL顯影液檢測,用Tanon 5200化學發(fā)光成像系統(tǒng)捕獲信號,曝光時間由機器自動設定。拍攝后,利用ImageJ軟件計算目的條帶灰度值,并利用GraphPad軟件分析數據及制作柱狀圖。
2 結果
2.1變異檢測及驗證結果 通過軟件分析及數據過濾,將測序的原始數據進行去接頭、去重復,過濾低質量數據,3例患者樣本的數據量、測序深度及覆蓋度見表1。
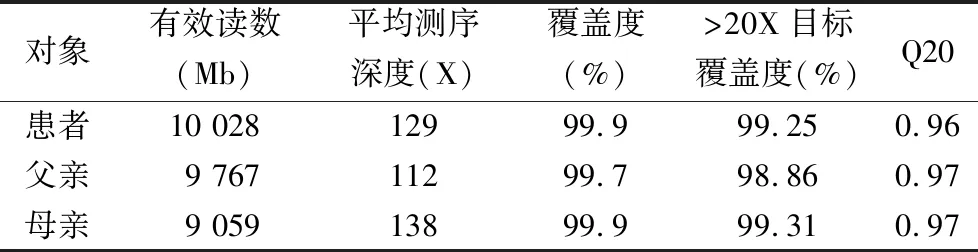
表1 3例研究對象樣本的測序數據量
進一步采用全外顯子測序檢測發(fā)現(xiàn),該患者16號染色體p11.2區(qū)域PRRT2基因的第2個外顯子編碼區(qū)檢測到1個雜合的無義突變(Chr16:29825024 C>T;c.649C>T;p.R217X, NM_145239.3),遺傳自母親。根據美國醫(yī)學遺傳學與基因組學學會(ACMG)原則(2015)[4]對該變異進行評估,其變異分類聯(lián)合標準評分為PVS1+PS1+PM1+PM2+PP4,綜合評定為致病性突變。用Sanger測序對外顯子測序結果進行驗證,結果證實該患者的無義突變c.649C>T來自于母親。見圖1。
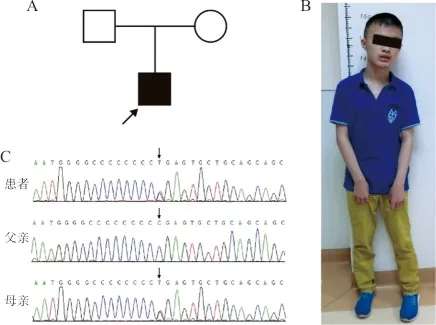
注:A,該患者家系圖;B,該患者的臨床特征;C,Sanger測序驗證患者及母親PRRT2基因突變。箭頭表示點突變所在位置及堿基,患者及母親均存在雜合突變。
2.2R217X突變導致PRRT2蛋白被過早降解 野生型及突變質粒轉染HEK293T細胞后,經western blot分析結果顯示,表達突變型Prrt2的細胞無法檢出Prrt2(識別蛋白氨基端)和FLAG(識別蛋白羧基端)信號,Prrt2 R223X不能表達,提示PRRT2 R217X在患者體內不能正常表達。見圖2。
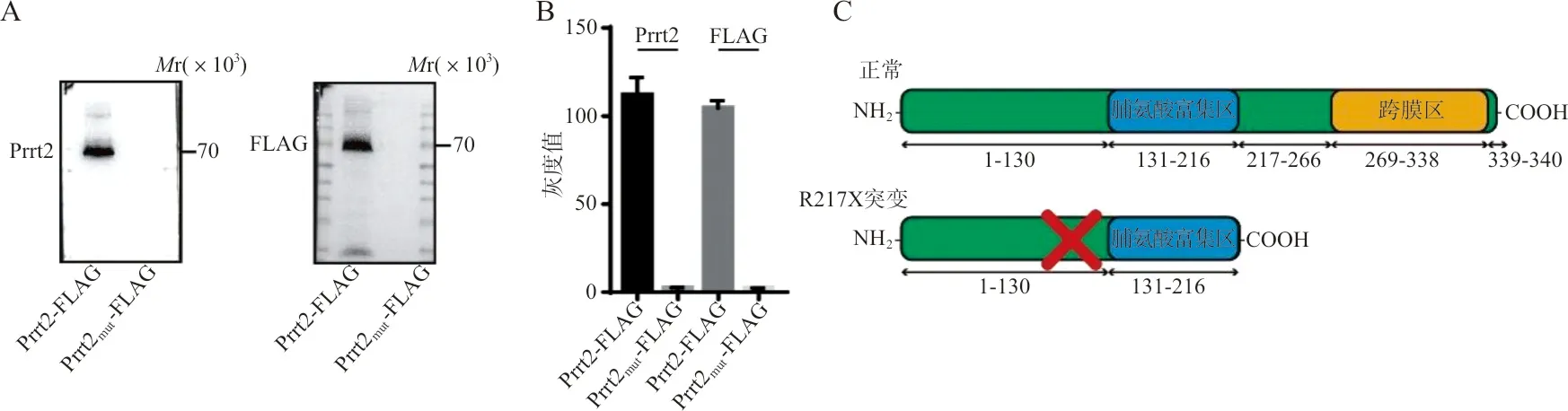
注:A,western blot條帶圖,Prrt2為使用anti-Prrt2抗體溫育,F(xiàn)LAG為使用anti-FLAG抗體溫育,Prrt2-FLAG為野生型蛋白,Prrt2mut-FLAG為R223X突變蛋白;B,western blot條帶的灰度值統(tǒng)計柱狀圖;C,人PRRT2 R217X突變與正常PRRT2蛋白表達異常對比的示意圖。
3 討論
在本研究中,我們通過全外顯子測序技術鑒定了1例智力障礙合并癲癇及運動障礙的男性患者的PRRT2基因存在雜合無義突變(c.649C>T;p.R217X)。PRRT2基因(NM_145239.2)位于16p11.2,含有4個外顯子,編碼340個氨基酸。該基因最常見的突變?yōu)閏.649dupC,與本研究發(fā)現(xiàn)的突變相似,均導致編碼蛋白質在脯氨酸富集區(qū)與跨膜區(qū)之間提前終止翻譯。目前,該基因突變被報道的主要臨床癥狀為癲癇及運動障礙,這與該患者的部分臨床癥狀吻合[5-6]。據文獻報道,該基因翻譯蛋白PRRT2通過參與形成突觸前膜神經遞質,釋放復合物,負向調控神經遞質釋放[7]。因此,突變導致調控機制失調,后膜興奮性異常增高,最終引發(fā)運動障礙以及癲癇。然而,最近的研究發(fā)現(xiàn),一些PRRT2突變患者表現(xiàn)出非典型癥狀如偏頭痛和智力障礙[8-9],但原因未明。目前,僅少數病例報道為智力障礙表型[4]。攜帶純合PRRT2截斷突變的患者中有52.4%(11/21)存在智力障礙,而攜帶雜合突變的患者中只有0.6%(8/1 423)存在智力障礙。PRRT2突變的智力障礙患者在嬰幼兒時期往往出現(xiàn)嚴重的癲癇甚至強直性陣攣發(fā)作[10],可能引起腦組織損傷從而影響智力。然而,本研究中的病例沒有嚴重的癲癇大發(fā)作并且腦CT及腦電圖均無異常,提示該患者的智力障礙與重度癲癇之間可能無必然聯(lián)系。值得注意的是,患者母親也是該雜合突變的攜帶者,但無任何臨床癥狀。曾有報道PRRT2突變家系中存在外顯不全的個體[11]。因此,推測患者母親可能同樣為PRRT2突變不完全外顯。
PRRT2在哺乳動物中高度保守,人和小鼠PRRT2蛋白脯氨酸富集區(qū)至跨膜區(qū)氨基酸序列完全一致,小鼠Prrt2的Arg223氨基酸位點對應人PRRT2基因的Arg217位點。為了進一步研究PRRT2(c.649C>T)的突變致病機制,我們克隆了小鼠Prrt2基因并設計了R223X(c.667C>T)突變模擬人類PRRT2_R217X突變。結果發(fā)現(xiàn),Prrt2_R223X蛋白不能正常表達,提示PRRT2_R217X突變患者PRRT2蛋白體內異常表達,且低于健康人,這與既往報道的PRRT2R217X附近的截斷突變導致蛋白質過早被降解相一致[12]。本研究局限性在于PRRT2蛋白僅在腦組織中特異表達,人體樣本取材困難,我們僅從體外驗證PRRT2 R217X蛋白質表達異常,暫時無法進行體內實驗驗證缺少PRRT2是否影響智力發(fā)育。隨著科技水平進步,相關終會揭示其更深層的致病機理并以此治療此類患者。
綜上所述,本研究發(fā)現(xiàn)PRRT2基因截斷突變除了會導致癲癇、運動障礙等常見臨床癥狀外,還會引起智力障礙等神經系統(tǒng)發(fā)育異常。檢測PRRT2基因對于一些同時存在智力發(fā)育障礙以及運動系統(tǒng)障礙的患者具有一個預警的作用。外顯子測序技術可以快速、準確地診斷該類患者的遺傳學病因。
猜你喜歡
中國民間療法(2021年5期)2021-06-09 09:21:04
中華養(yǎng)生保健(2020年2期)2020-11-16 00:49:00
解放軍醫(yī)學院學報(2020年12期)2020-03-29 05:11:46
中成藥(2017年6期)2017-06-13 07:30:35
飲食科學(2017年5期)2017-05-20 17:11:53
臨床醫(yī)藥文獻雜志(電子版)(2017年11期)2017-05-17 04:48:10
安徽醫(yī)科大學學報(2015年9期)2015-12-16 11:09:44
中國當代醫(yī)藥(2015年7期)2015-03-01 02:01:13
西南軍醫(yī)(2015年4期)2015-01-23 01:19:30
西部中醫(yī)藥(2014年6期)2014-03-11 16:07:47
