POR基因突變致先天性腎上腺皮質增生癥兩家系遺傳學分析
2020-09-17 08:50:38李莉劉玲毛會英
海南醫學 2020年17期
李莉,劉玲,毛會英
1.瀘州市婦幼保健院兒科,四川 瀘州 646000;2.廣東醫科大學附屬第三醫院兒科,廣東 佛山 528000
p450 氧化還原酶(p450 oxidoreductase,POR)基因位于7號常染色體長臂(Chr.7q11.2),長約7.1 kb,共16個外顯子,編碼680 個氨基酸組成黃素蛋白(NCBI Reference Sequence: NG_008930.1)[1],并參與電子傳遞反應。POR基因突變可影響21-羥化酶和17α-羥化酶的活性而導致p450 氧化還原酶缺陷癥(p450 oxidoreductase deficiency,PORD)[2],其臨床主要表現為骨骼發育不良(skeletal dysplasia)、腎上腺功能紊亂(adrenal dysfunction)、性發育異常(disorders of sex development,DSD)以及母孕期的女性男性化[3-5],先天性腎上腺皮質增生癥(congenital adrenal hyperplasia,CAH)是少見類型之一,為常染色體隱形遺傳病[6],目前對PORD 病例報道較少。本研究對21-羥化酶A2、11-羥化酶B1 及17-羥化酶A1 基因無異常的CAH 患者進行POR 基因檢測,為臨床診斷提供依據,提供遺傳咨詢。
1 資料與方法
1.1 一般資料 選取2019年1~12月瀘州市婦幼保健院及廣東醫科大學附屬第三醫院兒科門診確診的CAH患兒,收集21-羥化酶A2、11-羥化酶B1及17-羥化酶A1基因正常的2例患兒臨床資料,并對其家系進行POR基因檢測。
1.2 方法 經家長知情同意并簽字后,留取患兒及其父母靜脈血2 mL。提取全血基因組DNA 和RNA,設計14 對引物對POR 基因的16 個外顯子及其側翼進行PCR 擴增后Sanger 測序,上下游引物如表1所示。結果與GenBank中POR基因序列進行比對,并在其家屬中進行驗證。根據突變位置設計RNA引物,進行RNA逆轉錄后進行測序,測序結果在GenBank中進行比對,進一步驗證突變位點,RNA 引物:For1,5'-CCACCTCATGCACCTGGAAT-3';Rev1,5'-CCCGGTACAGGTAGTCCTCA-3'。

表1 POR基因各外顯子PCR擴增引物
2 結果
2.1 臨床資料 先證者1,男性,1個月13 d,以體質量不增為首發癥狀,起病時間早,伴嘔吐、腹瀉,查體發現生殖器畸形(尿道下裂-會陰型),全身皮膚輕微色素沉著,雙側陰囊色素沉著明顯(圖1 A),輔助檢查:電解質提示低鈉高鉀,17OHP 升高、醛固酮降低等(表2),染色體檢查為正常男性核型。先證者2,男性,2歲11個月,以“陰莖短小”為主要表現(圖1 B),無腹瀉、嘔吐、體質量不增等癥狀,查體發現陰莖外觀短小,總長約4.5 cm,包埋部分約2.0 cm,陰莖彎曲,睪丸容積較同齡兒偏大(表2),輔助檢查見表3。
2.2 基因檢測結果
2.2.1 先證者1 外顯子13,c.1508C>T (純合突變),該突變導致POR 第503 位丙氨酸突變為纈氨酸(Valin,V/Val,GCC→GTC,p.A503V),如圖2。該突變遺傳自先證者1父母。
2.2.2 先證者2 外顯子15,c.1820A>G (純合突變),導致POR 第607 位氨基酸由酪氨酸(tyrosine,Y/Tyr)突變為半胱氨酸(cysteine,C/Cys,TAC→TGC,p.Y607C),如圖3。該突變遺傳自先證者2父母。
2.2.3 POR基因突變 POR基因外顯子13,c.1508C>T 及外顯子15,c.1820A>G 經Alamut Visual 及Mutatior Taster 功能軟件預測會影響蛋白結構域的功能,通過Phyre2 功能軟件可知503 位丙氨酸及607 位酪氨酸位于相對保守區域,如圖4。
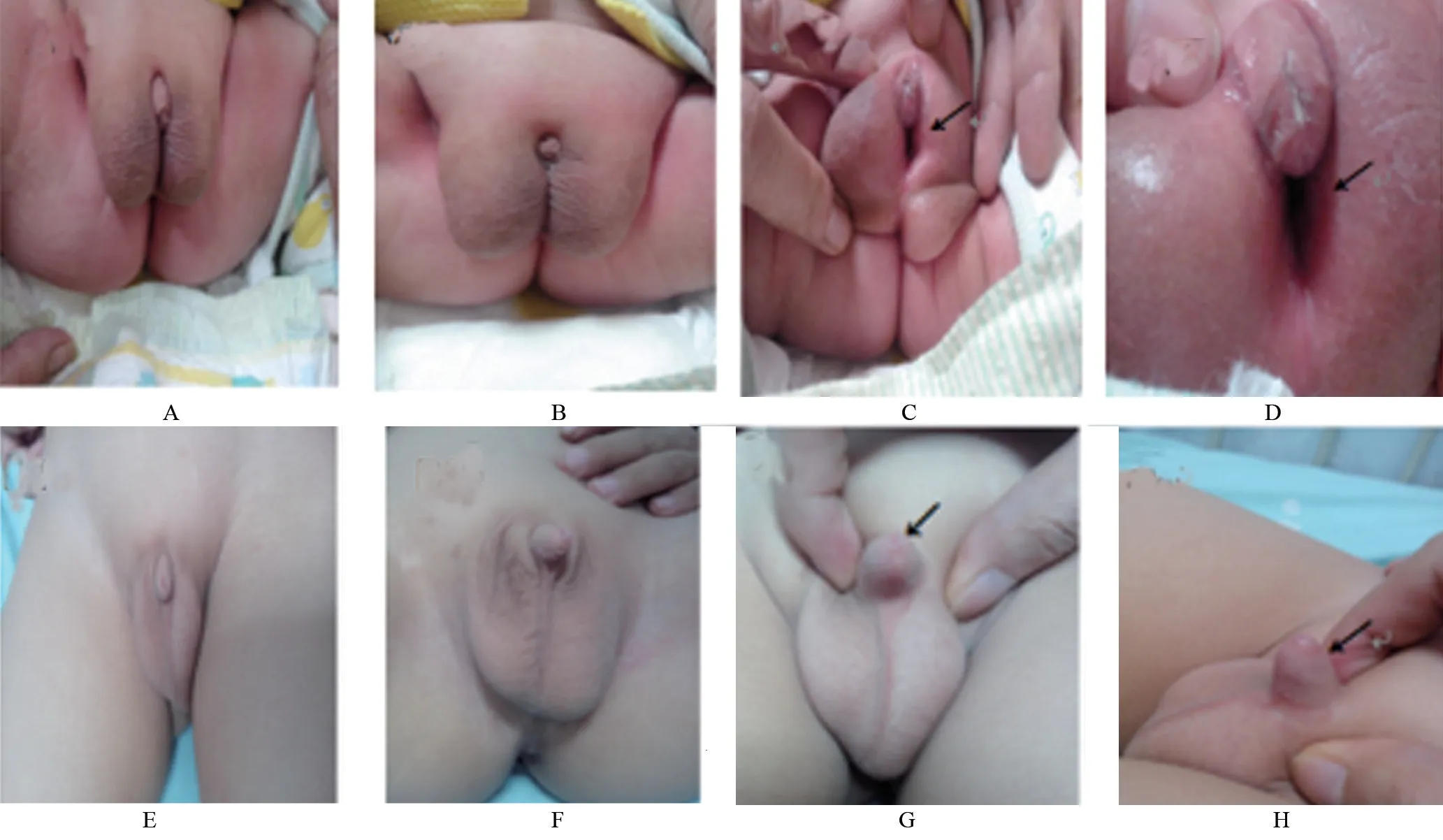
圖1 兩位先證者生殖器檢查

表2 兩位先證者的實驗室檢查
2.3 治療 先證者1 給予糖皮質激素(氫化可的松片)、鹽皮質激素(9α-氟氫化可的松)及氯化鈉治療,2 周后血鈉、血鉀恢復正常;先證者2給予糖皮質激素(氫化可的松片)治療。
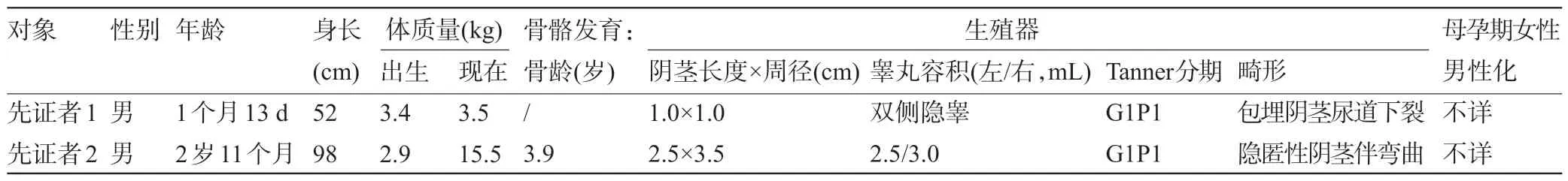
表3 兩位先證者的臨床評估
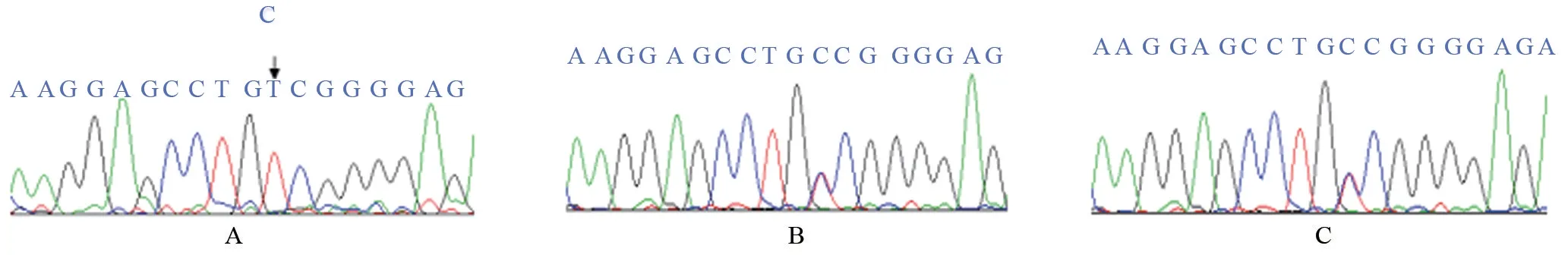
圖2 外顯子13基因測序圖
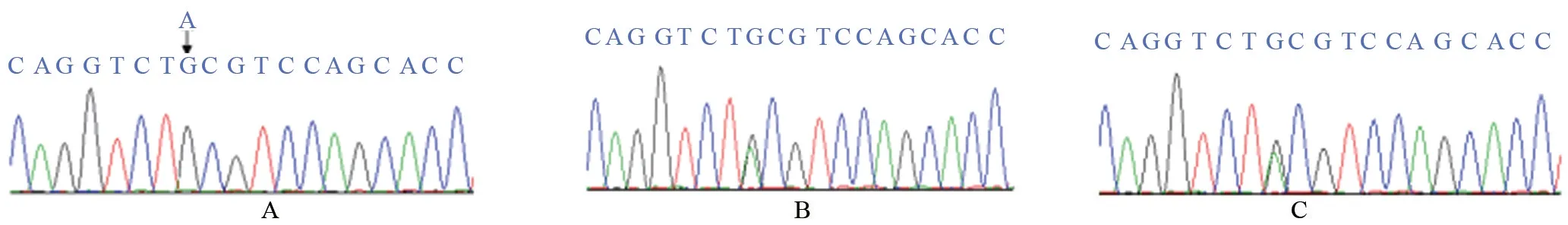
圖3 外顯子15基因測序圖
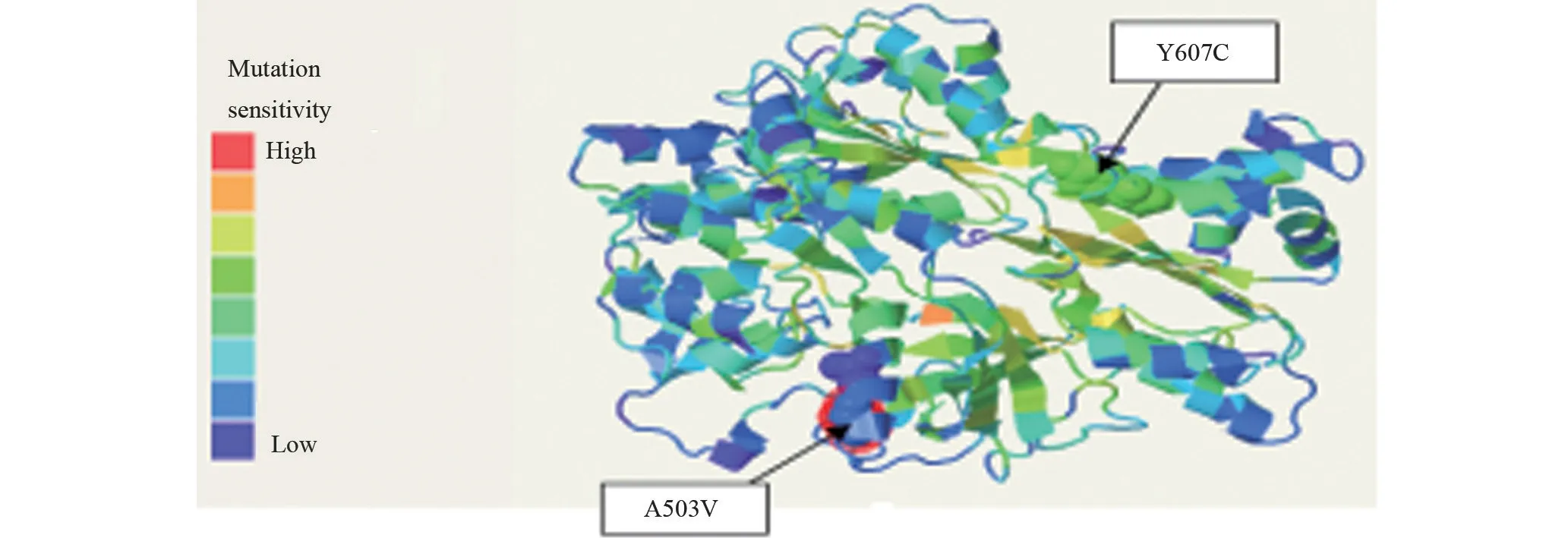
圖4 POR突變圖
3 討論
CAH 是由于編碼腎上腺皮質激素合成酶的一組基因發生突變,導致酶缺陷繼而引起皮質激素合成不足,繼發垂體促腎上腺皮質激素(adreno-cortico-tropic-hormone,ACTH)和下丘腦促腎上腺皮質激素釋放激素(corticotropin releasing hormone,CRH)分泌增加,最終導致腎上腺皮質增生的常染色體隱形遺傳病。CAH除了常見的21-羥化酶缺乏癥(21-OHD)外,還有11α-羥化酶缺乏癥(11α-OHD)、17-羥化酶缺乏癥(17-OHD)、類脂性腎上腺皮質增生癥(StAR)和p450氧化還原酶缺陷癥(PORD)等少見類型。POR 是內質網中類固醇和藥物代謝細胞色素p450 蛋白的代謝反應所必需的酶,是兩種主要的類固醇生成酶17-羥化酶和21-羥化酶的電子供體[7]。POR突變使p450酶系活性受到影響,最終影響藥物的代謝及類固醇激素的合成,導致一系列復雜的疾病,類似于多種類固醇代謝酶的聯合缺陷[8-9]。
本研究中先證者1臨床表現及實驗室檢查結果類似21-OHD 經典型,但21-羥化酶A2 基因無異常。POR基因提示外顯子13,c.1820A>G(p.A503V,純合突變),該突變來自先證者父母(雜合突變)。該位點為POR基因常見突變位點,影響類固醇合成酶及藥物代謝酶活性。研究發現,c.1820A>G 突變使17-羥化酶與21-羥化酶活性分別降低32%與20%,并使17、20裂解酶活性降低42%[10-11],導致皮質醇和醛固酮合成受損,皮質醇低下,經負反饋使ACTH分泌增加,刺激腎上腺皮質細胞增生,以增加皮質醇合成,但17-羥化酶與21-羥化酶活性降低使皮質醇依然低下,且孕酮轉化為醛固酮降低,導致水鹽平衡失衡;但雄激素合成通路正常,在高ACTH刺激下,堆積的17-OHP和孕酮向雄激素轉化增多,產生了旁路代謝亢進-高雄激素血癥(雄烯二酮、睪酮和脫氫表雄酮升高),臨床表現與21-OHD 相似。先證者2,外顯子15,c.1820A>G (p.Y607C,純合突變),該突變來自先證者父母(雜合突變)。POR 第607 位酪氨酸位于煙酰胺腺嘌呤二核苷酸磷酸(NADPH)結合域,酪氨酸的羥基和NADPH 第一正磷酸基團之間形成氫鍵,使NADPH 和黃素腺嘌呤二核苷酸(FAD)緊密結合,傳遞電子[12-13]。POR 第607 位酪氨酸突變為半胱氨酸改變了NADPH 與FAD的結合,從而使POR活性下降60%~90%[14-15],抑制17-羥化酶、21-羥化酶與19-羥化酶活性,但以19-羥化酶為主,皮質醇和醛固酮通路基本正常,睪酮轉化為雌二醇通路受阻,使其不能轉化為雌二醇,導致高雄激素血癥,因此先證者2 臨床表現較先證者1 輕。本研究兩位先證者均為純合突變,突變均來源其雜合突變的父母,符合常染色體隱性遺傳規律。
PORD 的臨床表現聯合了21-OHD 和17-OHD,根據不同突變對酶活性的影響程度,PORD的臨床表現多樣,容易導致臨床上CAH患者由于臨床表現、體征和實驗室檢查不典型而出現誤診漏診。PORD 治療同21-OHD,最重要的是糾正腎上腺皮質功能減退危象,維持機體正常的生理代謝,降低死亡率并且抑制垂體ACTH 分泌,延緩腎上腺雄激素過度分泌導致的性早熟。治療不當與治療過度均可導致矮小及生理、心理發育障礙等后遺癥。因此定期隨訪很重要,根據臨床癥狀及檢查結果及時調整治療方案,以最低藥物劑量達到最好的代謝控制,避免或減少藥物副作用,使患者能達到正常的生長及青春發育,提高生活質量。
本研究通過對2例PORD患兒的臨床表現和基因突變特點分析,POR 基因外顯子13,c.1508C>T、外顯子15,c.1820A>G突變是這兩例患者的致病原因,提高了臨床醫師對本病的認識,并擴大了基因突變譜,為遺傳咨詢提供了基礎。
