托吡酯對酸敏感離子通道1a調控作用的研究
2018-03-07 09:41:10李雯,魏東
中風與神經(jīng)疾病雜志 2018年2期
李 雯, 魏 東
細胞外微環(huán)境pH值的改變對于神經(jīng)細胞發(fā)揮正常的生理功能是非常重要的[1]。在炎癥、缺血、創(chuàng)傷、退行性疾病以及癲癇發(fā)作等神經(jīng)系統(tǒng)病理情況下,厭氧糖代謝造成的乳酸堆積和ATP水解等釋放大量的H+使得組織內pH值極大地降低而出現(xiàn)組織酸化[2~9],直接或間接導致神經(jīng)元損傷及功能改變[10]。1997年,一種能夠被酸化所激活的陽離子通道-酸敏感離子通道(acid-sensing ion channels,ASICs)被發(fā)現(xiàn)之后[11],大量的研究發(fā)現(xiàn)ASICs特別是其1a亞型(ASIC1a)廣泛表達于中樞神經(jīng)系統(tǒng)且在神經(jīng)系統(tǒng)的生理和病理過程中發(fā)揮著重要的作用[12],尤其是在癲癇的發(fā)生及發(fā)展領域,ASIC1a已經(jīng)成為一個研究的熱點。托吡酯(topiramate,TPM)作為選擇性電壓依賴的鈉離子通道阻斷劑,是否通過對ASICs的調節(jié)而發(fā)揮抗癲癇作用尚無研究報道,因此,我們采用全細胞記錄膜片鉗及Western blot定量分析TPM對ASICs介導電流的調控作用及對其蛋白表達的影響,來探討ASICs是否是TPM抗癲癇治療的作用靶點。
1 材料與方法
1.1 材料 (1)實驗動物:懷孕16~17 d的雌性SD大鼠(第四軍醫(yī)大學動物中心);(2)實驗細胞及質粒:CHO細胞及ASIC1a、ASIC2a及ASIC3 3種質粒(上海交大醫(yī)學院徐天樂老師惠贈);(3)主要試劑:F12培養(yǎng)基、L型谷氨酰胺等細胞培養(yǎng)相關試劑(美國GIBCO公司),HilyMax質粒轉染試劑(日本Dojindo Laboratiories公司),BCA試劑盒(美國Pierce公司),ASIC1a抗體(美國Milipore公司),辣根過氧化物酶標記的二抗(美國Milipore公司),GAPDGH抗體(美國CST公司),Nacl、Kcl等電生理試劑(美國Sigma公司)。
1.2 胞系的培養(yǎng)及傳代 CHO細胞培養(yǎng)在37 ℃、5% CO2孵育箱中內,采用F12培養(yǎng)基加入50 U/ml的青霉素、1 mmol/L L型谷氨酰胺、50 μg/ml的鏈霉素和10%的胎牛血清,觀察細胞生長情況,每2~3 d半量換液。在無菌情況下傳代,先吸棄原有培養(yǎng)基,PBS洗3遍,加入0.125%的胰蛋白酶消化2~3 min,然后加入含血清的F12培養(yǎng)基終止消化,將貼壁細胞吹打脫落,吹散后進行離心5 min(1000 r/min),棄上清后,再加入適量培養(yǎng)基,充分吹打重懸,計數(shù)后進行接種備用。
1.3 大鼠皮質神經(jīng)元原代培養(yǎng) 將懷孕16~17 d的雌性SD大鼠進行乙醚氣體麻醉,剪開消毒過的腹壁并暴露子宮,取6~8個胚胎置于玻璃皿中。無菌的情況下從子宮中剖取胎鼠后夾取頭部置于預冷過的DMEM溶液中,解剖顯微鏡下充分剝離腦膜,剝去大腦皮質以外的其它結構。將其剪碎后,加入0.05%胰蛋白酶于37 ℃、5% CO2孵育箱中消化約5 min,加入10%的胎牛血清高糖DMEM溶液終止消化,充分吹打后過濾、1000 r/min,5 min離心,棄上清后加入適量接種培養(yǎng)基充分吹打重懸,計數(shù)后進行接種備用。次日更換半量維持培養(yǎng)基(含2%的B27及1 mmol/L L型谷氨酰胺的Neural basal培養(yǎng)液),以后根據(jù)細胞生長情況約2~3 d半量更換培養(yǎng)基。
1.4 細胞轉染 培養(yǎng)的CHO細胞融合至70%~80%時進行脂質體轉染,實驗方案根據(jù)HilyMax liposome transfection reagent所提供的操作方法進行。在無菌情況下操作,每個60 mm培養(yǎng)皿加入約7~9 μg質粒;每個35 mm培養(yǎng)皿加入約3~4 μg質粒。轉染4 h后吸棄含脂質體的培養(yǎng)基,PBS洗一次后更換為維持培養(yǎng)基;轉染后的24~48 h內進行蛋白質提取以及電生理實驗。當進行兩種不同的質粒共轉染時,采用相同的質粒量進行配比混合。
1.5 電生理記錄 本實驗電生理采用電壓鉗條件下的全細胞記錄模式。配置電極內液及細胞外液的,玻璃微電極采用兩步垂直拉制方法,使其入水電阻約為3.5~5.5 M。灌流給藥采用“Y”管系統(tǒng)依靠重力實現(xiàn)[13]。實驗時首先給予玻璃電極內施加正壓,通過步進式微操儀將玻璃電極尖端輕壓在細胞表面,撤除正壓,使細胞膜吸附于電極尖端,給予串聯(lián)電阻的自動補償,逐漸給予負壓吸引,施加-60 mV鉗制電壓,待形成超過G級別的巨阻抗后用負壓將細胞膜吸破,使細胞內液與電極內液互通而形成全細胞記錄。所記錄信號通過放大器采集后通過數(shù)模轉換器輸入計算機,使用pCLAMP 9.0軟件進行數(shù)據(jù)采集、設置調整,使用Clampex 9.0進行數(shù)據(jù)分析。
1.6 免疫印跡
1.6.1 樣本制備 將神經(jīng)元接種于60 mm培養(yǎng)皿中,培養(yǎng)6~7 d后給于TPM藥物處理24 h,加入RIPA裂解液,冰上刮取細胞后進行冰上裂解15 min,收集裂解液置入EP管中,進行4 ℃低溫離心(13000 r/min)20 min,收集上清,加入SDS上樣緩沖液,金屬浴(95 ℃)煮沸5 min,置于-80 ℃保存。
1.6.2 電泳、轉模及免疫顯色檢測 首先制備SDS PAGE膠進行電泳,結束后用半干轉膜法將蛋白樣品從分離膠中轉移至PVDF膜上,將PVDF膜在20%脫脂奶粉PBST溶液中室溫封閉1 h,然后加入抗體稀釋液配制的一抗(1∶1000),4 ℃條件下旋轉孵育過夜,加入稀釋配制的二抗(1∶2000),室溫下孵育2 h,最后在暗室中進行ECL顯色,使用X光膠片顯影,膠片掃描后使用Image Pro Plus 6.1軟件進行圖像分析和數(shù)據(jù)采集。
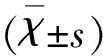
2 結 果
2.1 TPM對不同ASIC亞單位的調控作用 根據(jù)神經(jīng)系統(tǒng)ASICs的表達模式,在CHO細胞系上分別轉染了ASIC1a、ASIC2a、ASIC1a+ASIC2a和ASIC3質粒,得到ASIC1a同聚體、ASIC2a同聚體、ASIC1a/ASIC2a異聚體和ASIC3同聚體通道。分別對這4種組合進行電生理記錄并給予TPM(300 μM)(見圖1),發(fā)現(xiàn)不同亞單位組成的離子通道其所通透的內向電流曲線對TPM的反應不同,TPM僅對ASIC1a亞單位組成的同聚體通道有增強調控作用,而對另外3種通道均無明顯的改變。
2.2 TPM對ASIC1a同聚體通道調節(jié)作用 TPM對ASIC1a同聚體通道介導的峰電流的增強效應也具有劑量依賴的特性,劑量越大,效應越大,最大可有近60%的增強效應,其半數(shù)有效劑量約為60 μM(見圖2A)。這種增強效應與胞外酸化程度相關,100 μM的TPM可以增強18%左右pH 6.8誘導的峰電流強度,而在pH 6.0時則約為40%(見圖2B)。隨后我們采取了4種不同的TPM給藥方式:僅與酸共給、僅預給10 s、僅預給10 min、先預給10 s然后與酸共給,發(fā)現(xiàn)必須在與酸共給的情況下才具有增強效應,而兩種僅預給的方式都不能產(chǎn)生增強效應,這就提示TPM的作用具有通道狀態(tài)依賴的特性,只有通道被打開后才能發(fā)揮作用(見圖2C、D)。
2.3 TPM對ASIC1a蛋白的表達作用 我們給于原代培養(yǎng)的神經(jīng)元不同濃度的TPM作用24 h后提取蛋白樣品,通過Western blot定量分析,發(fā)現(xiàn)TPM并不影響ASIC1a的蛋白表達(見圖3A、B)。

圖1 托吡酯對不同ASICs通道內向電流的調控作用
(注:n>6,與給藥前相比*P<0.05,NS表示無統(tǒng)計學差異)

圖2 托吡酯對ASIC1a同聚體通道內向電流的調控作用
(注:n>6, 與給藥前相比*P<0.05 ,NS表示無統(tǒng)計學差異)

圖3 托吡酯對ASIC1a的蛋白表達的影響
(注:n=4,NS表示無統(tǒng)計學差異)
3 討 論
酸敏感離子通道1a(acid-sensing ion channel 1a,ASIC1a)作為中樞神經(jīng)系統(tǒng)內表達最廣泛的ASICs家族成員之一,因其分布廣泛、激活閾值較低并能通透鈣離子而受到研究者的重視,當大腦皮質和海馬神經(jīng)元中ASIC1基因被敲除后,原來酸所激活的電流幾乎被全部去除[14~16],提示ASIC1a是中樞系統(tǒng)神經(jīng)元中ASICs家族主要的功能性亞單位。大量的研究發(fā)現(xiàn)ASIC1a參與了體內眾多的生理及病理過程。已經(jīng)發(fā)現(xiàn)目前常用的各類藥物中非甾體抗炎藥[17]、麻醉劑利多卡因[18]以及治療多發(fā)性硬化的藥物4-氨基吡啶[19]對ASIC1a的功能具有調節(jié)作用,ASIC1a很可能作為這些藥物的靶點參與了疾病的治療過程之中。那么,托吡酯(topiramate,TPM)作為一種選擇性阻斷電壓依賴的鈉離子通道藥物,在抗癲癇治療的過程中是否也通過ASIC1a發(fā)揮作用的目前尚未報道,本實驗通過全細胞記錄模式及Western blot定量分析探討了TPM對ASIC1a電流和蛋白表達的調控作用,目的在于確定ASICs是否是TPM治療癲癇的可能作用靶點。
有研究發(fā)現(xiàn)ASIC1a非特異性抑制劑阿米洛利能夠延長由鋰-匹羅卡品誘導大鼠癲癇模型發(fā)作的潛伏期時間,提示ASIC1a可能是癲癇治療的重要靶點,但ASIC1a參與癲癇發(fā)作終止的機制尚不明確。有文獻報道癲癇發(fā)作后腦組織中出現(xiàn)明顯的酸中毒,導致組織內環(huán)境pH值下降,繼而激活了抑制性中樞神經(jīng)元上ASIC1a通道,使癲癇發(fā)作終止[20]。也有研究者發(fā)現(xiàn)在小鼠的癲癇模型中干擾ASIC1a表達后,增加了小鼠癲癇發(fā)作的嚴重程度,而在ASIC1a過表達小鼠中出現(xiàn)了相反的結果,這提示可能是由于內環(huán)境的酸化激活ASIC1a而興奮了抑制性中間神經(jīng)元,從而終止了癲癇發(fā)作[21]。
本研究結果顯示,TPM對ASIC1a同聚體通道產(chǎn)生的電流具有增強效應,而對ASICs家族其它3種亞型均無明顯的影響,這種增強效應呈現(xiàn)劑量依賴的特性,隨著藥物濃度的增大,增強的強度也逐漸增大,同時與酸化的程度也呈正相關。那么TPM就有可能通過增強ASIC1a的電流而使抑制性的中間神經(jīng)元興奮,最終實現(xiàn)對癲癇發(fā)作的抑制。因此ASIC1a很可能是TPM抗癲癇作用的靶點之一。
癲癇發(fā)作和癲癇發(fā)生是兩種不同的病理生理過程,我們的研究發(fā)現(xiàn)TPM對ASIC1a同聚體通道產(chǎn)生的電流具有增強效應而推論ASIC1a可能是TPM抑制癲癇發(fā)作的靶點之一,但在神經(jīng)系統(tǒng)廣泛表達的ASIC1a在癲癇發(fā)生的病理過程之中扮演什么樣的角色,仍然是未知的領域,需要進一步的研究去探索和明確。
綜上所述:我們發(fā)現(xiàn)TPM僅能增強ASICs家族中ASIC1a組成的同聚體通道所產(chǎn)生的內向電流,而對另外3種通道均無明顯的調控作用;這種增強效應具有劑量依賴的特性,劑量越大,效應越大;并且必須在與酸共給的情況下才能發(fā)揮增強效應;TPM并不影響ASIC1a的蛋白表達。這提示ASIC1a可能是TPM抑制癲癇發(fā)作的作用靶點之一。
[1]Chesler M.The regulation and modulation of pH in the nervous system[J].Prog Neurobiol,1990,34(5):401-427.
[2]Chesler M.Regulation and modulation of pH in the brain[J].Physiol Rev,2003,83(4):1183-1221.
[3]Kraut JA,Madias NE.Metabolic acidosis:pathophysiology,diagnosis and management[J].Nat Rev Nephrol,2010,6(5):274-285.
[4]Sutherland SP,Cook SP,McCleskey EW.Chemical mediators of pain due to tissue damage and ischemia[J].Prog Brain Res,2000,129:21-38.
[5]Somjen GG.Acidification of interstitial fluid in hippocampal formation caused by seizures and by spreading depression[J].Brain Res,1984,311(1):186-188.
[6]Wang RI,Sonnenschein RR.PH of cerebral cortex during induced convulsions[J].J Neurophysiol,1955,18(2):130-137.
[7]Messier C,Gagnon M.Glucose regulation and cognitive functions:relation to Alzheimer’s disease and diabetes[J].Behav Brain Res,1996,75(1/2):1-11.
[8]Bowen BC,Block RE,Sanchez-Ramos J,et al.Proton MR spectroscopy of the brain in 14 patients with Parkinson disease[J].AJNR Am J Neuroradiol,1995,16(1):61-68.
[9]Koga K,Mori A,Ohashi S,et al.H MRS identifies lactate rise in the striatum of MPTP-treated C57BL/6 mice[J].Eur J Neurosci,2006,23(4):1077-1081.
[10]Chesler M,Kaila K.Modulation of pH by neuronal activity[J].Trends Neurosci,1992,15(10):396-402.
[11]Waldmann R,Champigny G,Bassilana F,et al.A proton-gated cation channel involved in acid-sensing[J].Nature,1997,386(6621):173-177.
[12]Wemmie JA,Price MP,Welsh MJ.Acid-sensing ion channels:advances,questions and therapeutic opportunities[J].Trends Neurosci,2006,29(10):578-586.
[13]Li WG,Liu MG,Deng SN,et al.ASIC1a regulates insular long-term depression and is required for the extinction of conditioned taste aversion[J].Nat Commun,2016,7(7):13770.
[14]Wemmie JA,Chen J,Askwith CC,et al.The acid-activated ion channel ASIC contributes to synaptic plasticity,learning,and memory[J].Neuron,2002,34(3):463-477.
[15]Xiong ZG,Zhu XM,Chu XP,et al.Neuroprotection in ischemia:blocking calcium-permeable acid-sensing ion channels[J].Cell,2004,118(6):687-698.
[16]Askwith CC,Wemmie JA,Price MP,et al.Acid-sensing ion channel 2 (ASIC2) modulates ASIC1 H+-activated currents in hippocampal neurons[J].J Biol Chem,2004,279(18):18296-18305.
[17]Voilley N,de Weille J,Mamet J,et al.Nonsteroid anti-inflammatory drugs inhibit both the activity and the inflammation-induced expression of acid-sensing ion channels in nociceptors[J].J Neurosci,2001,21(20):8026-8033.
[18]Lin J,Chu X,Maysami S,et al.Inhibition of acid sensing ion channel currents by lidocaine in cultured mouse cortical neurons[J].Anesth Analg,2011,112(4):977-981.
[19]Boiko N,Kucher V,Eaton BA,et al.Inhibition of neuronal degenerin/epithelial Na+channels by the multiple sclerosis drug 4-aminopyridine[J].J Biol Chem,2013,288(13):9418-9427.
[20]Ziemann AE,Schnizler MK,Albert GW,et al.Seizure termination by acidosis depends on ASIC1a[J].Nat Neurosci,2008,11(7):816-822.
[21]Guo W,Chen X,He JJ,et al.Down-regulated expression of acid-sensing ion channel 1a in cortical lesions of patients with focal cortical dysplasia[J].J Mol Neurosci,2014,53(2):176-182.
猜你喜歡
核科學與工程(2021年4期)2022-01-12 06:30:26
中國民間療法(2021年5期)2021-06-09 09:21:04
今日農(nóng)業(yè)(2020年19期)2020-12-14 14:16:52
小學生必讀(中年級版)(2020年9期)2020-12-04 02:07:22
飲食科學(2017年5期)2017-05-20 17:11:53
中學物理·高中(2016年12期)2017-04-22 11:53:03
安徽醫(yī)科大學學報(2015年9期)2015-12-16 11:09:44
西南軍醫(yī)(2015年4期)2015-01-23 01:19:30
小櫻桃·童年閱讀(2014年11期)2014-12-01 22:21:30
中國中醫(yī)藥現(xiàn)代遠程教育(2014年20期)2014-03-01 04:31:21
