高效液相色譜-串聯質譜法測定鯉魚中硝基呋喃類代謝物的不確定度評估
2024-01-01 00:00:00王佳琪皇金龍林昕高顯穎劉利慧李劍軍
江西水產科技 2024年4期
關鍵詞:高效液相色譜


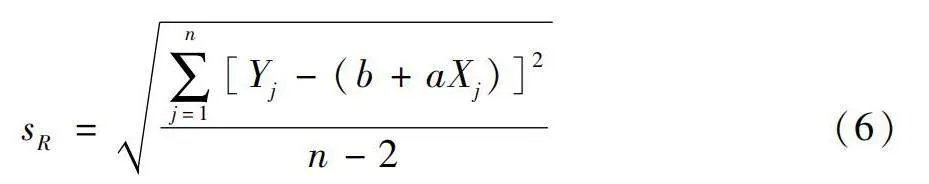

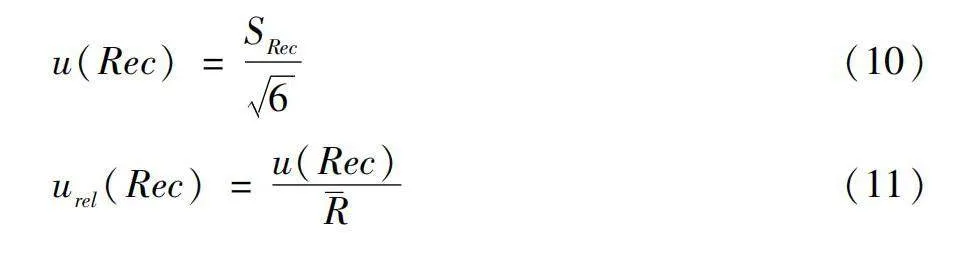
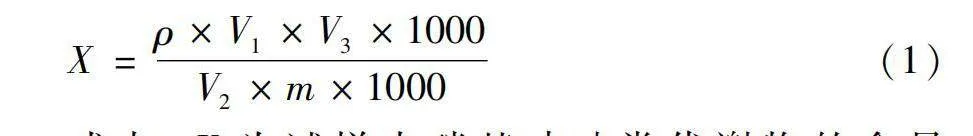

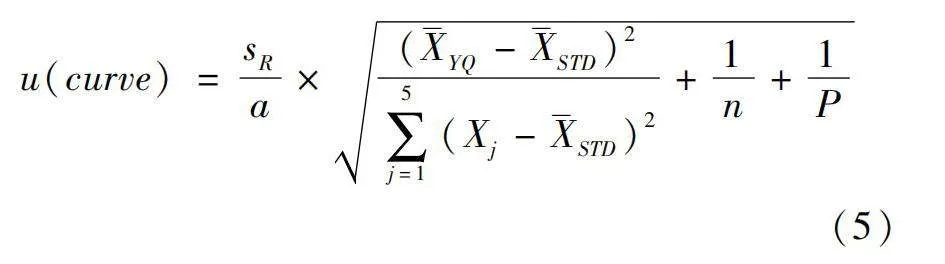


摘要:為應對自治區水產品中硝基呋喃類代謝物殘留檢測的能力驗證工作,保證基于國標方法的科學準確。通過建立數學模型對鯉魚中硝基呋喃類代謝物含量進行檢測,同時對不確定度進行評估。結果表明在6類主要不確定來源分量當中,隨機效應、標準溶液配制和加標回收為本實驗不確定度的主要來源。經評定在K=2情況下鯉魚中硝基呋喃類代謝物檢測含量為:AHD(5.157±0.631 0 μg/kg),AMOZ(4.813±0.487 7 μg/kg),AOZ(5.165±0.615 9 μg/kg),SEM(4.974±0.761 6 μg/kg)。
關鍵詞:高效液相色譜-串聯質譜法;硝基呋喃類代謝物;鯉魚;不確定度
中圖分類號:TS207 文獻標識碼:A
作者簡介:王佳琪(1992—),男,工程師,碩士,研究方向:農畜水產品藥物殘留檢測。E-mail:1396531031@qq.com
*通訊作者:李劍軍(1966—),男,研究員。E-mail:624062376@qq.com
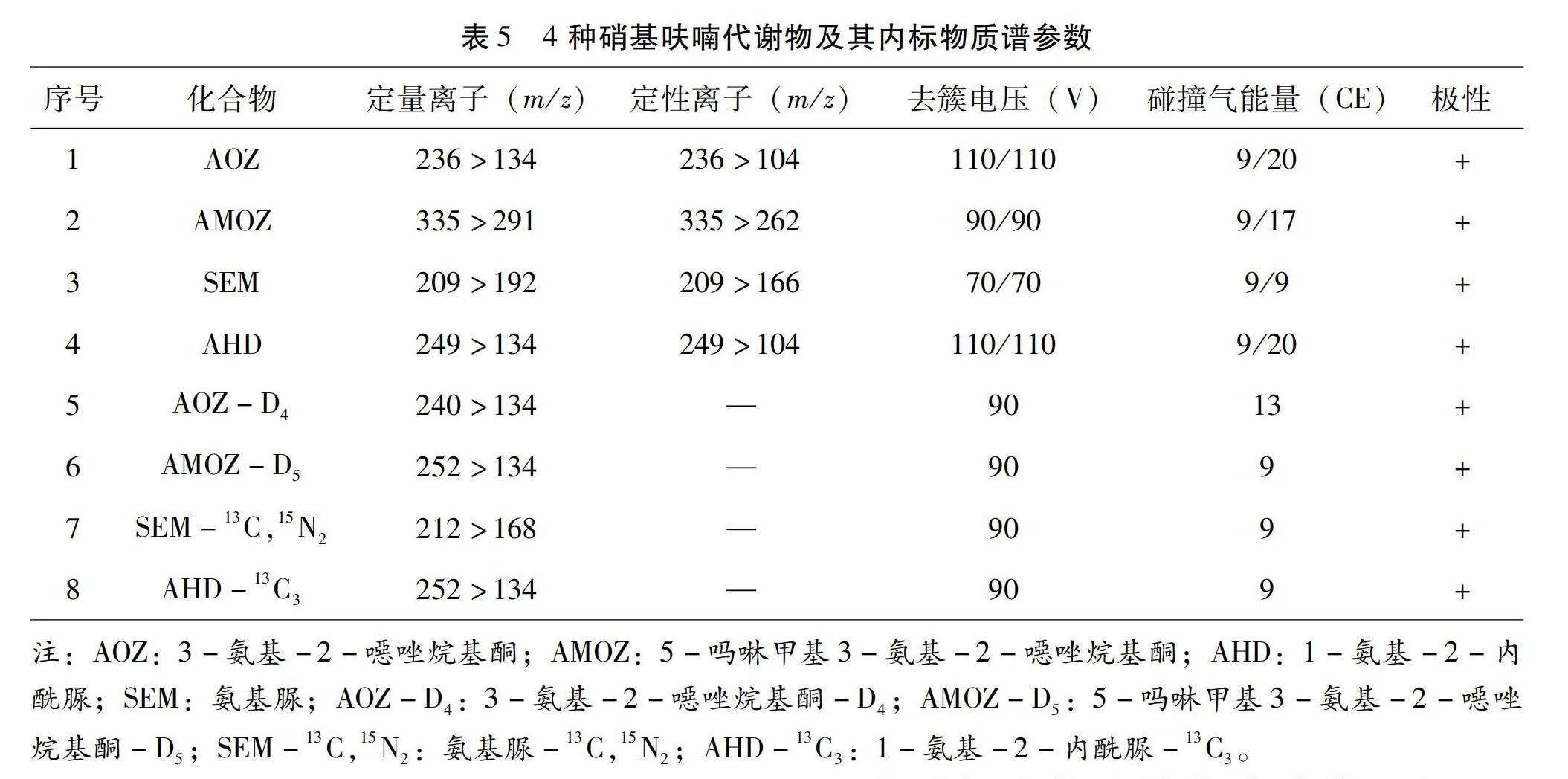
本文研究對象為4種硝基呋喃類代謝物,具體包括3-氨基-2-噁唑烷基酮(3-amino-2-oxazolidinone,AOZ)、5-嗎啉甲基3-氨基-2-噁唑烷基酮(5-morpholinomethyl-3-amino-2-oxazolidinone,AMOZ)、1-氨基-2-內酰脲(1-aminohydantoin,AHD)和氨基脲(semicarbazide,SEM),它們作為一類人工合成的廣譜類抗菌藥物被廣泛用于飼料添加和水產養殖過程中[1]。但是眾多研究表明,該類藥物具有非常強的致畸、致癌、致突變等毒副作用[2],并且由于其在環境和機體內半衰期長的特點[3-6],不僅對自然環境有害,并且嚴重威脅人類健康。因此,歐盟、日本和中國已經將硝基呋喃類代謝物作為禁用藥物被禁止使用,并將該類藥物作為動物源食品必檢項目列入檢測計劃。
硝基呋喃藥物原體極不穩定,在機體中通常短時間內被代謝,無法被及時采集并檢測,但其代謝物能夠與蛋白質穩定結合。因此,檢測硝基呋喃類代謝物可以間接代表硝基呋喃類原藥的殘留狀況[7]。當前,液相色譜(LC)和液相色譜串聯質譜(LC-MS/MS)法,作為比較常規的檢測硝基呋喃類代謝物的檢測手段,在基層檢測站和市縣級檢測中心應用非常普遍,但由于質譜法定性定量更準確、檢出限更低而成為該類藥物主流的檢測方法。不確定度通常用來衡量檢測過程中由于測量誤差的存在,造成的與被測量真實值之間的偏離程度,這種偏離程度一般用量值范圍來表示,通過這種方式來真實的反映被測量值的可信度。在檢測過程中不確定度會存在于每一個實驗步驟和所使用的實驗材料當中。因此,評估不確定度對于提高檢測結果可信度,以及檢測數值的判定具有現實意義。本文利用《測量不確定度評定與表示》(JJF 1059.1—2012)對基于《食品安全國家標準 水產品中硝基呋喃類代謝物多殘留的測定 液相色譜-串聯質譜法》(GB 31656.13—2021)的優化方法,在鯉魚硝基呋喃類代謝物殘留檢測的過程中進行不確定度的評定,旨在明確不確定度的主要來源,并在接下來自治區水產中硝基呋喃類代謝物藥物殘留檢測(盲樣考核)的檢測流程中有目的的降低有關不確定度的影響,從而保證檢測結果的可靠性。
1 材料與方法
1.1 材料與儀器
Agilent 1290-6470三重四極桿液相色譜-串聯質譜儀(美國安捷倫公司);3-18KS低溫高速離心機(德國Sigma Laborzentrifugen 公司);IKA HS 260 basic型震蕩儀(德國IKA公司);IKA MS 3 basic渦旋震蕩儀(德國IKA公司);N-EVAP 116氮吹儀(美國Organomation公司);TD-C系列電子天平(天津天馬衡基儀器有限公司);FiveEasy Plus FP20酸度計[梅特勒托利多科技(中國)有限公司]。
乙腈、甲醇、甲酸(色譜純,北京邁瑞達科技有限公司);純化水為屈臣氏蒸餾水(屈臣氏商標有限公司);0.22 μm微孔濾膜(上海安譜實驗科技股份有限公司)。
乙腈中4種硝基呋喃類代謝物混合標準溶液(100 mg/L)、甲醇中4種硝基呋喃類代謝物內標混合標準溶液(100 mg/L)含量均≥98.0%(購自天津阿爾塔科技有限公司)
1.2 實驗方法
1.2.1 前處理過程
取鯉魚背部肌肉500 g用組織勻漿機充分勻漿,于自封袋中-18 ℃避光保存。取2 g勻漿組織(精確到±0.01 g)于50 mL離心管中加入100 μg/L的內標工作液50 μL,渦勻,加入5 mL鹽酸溶液(0.5 mol/L)和150 μL的2-硝基苯甲醛溶液(0.05 mol/L),渦勻1 min,置于恒溫振蕩器中,37 ℃震蕩16 h。
離心管冷卻到室溫后,用磷酸氫二鉀溶液(1.0 mol/L)調節pH至7.0~7.5,再加入乙酸乙酯10 mL,渦勻2 min,10000 r/min離心5 min,準確分取5 mL上層有機相于玻璃管中,40 ℃水浴氮氣吹干,用1.0 mL的甲醇溶液(5%)溶解殘渣后,轉移至1.5 mL離心管中,在4 ℃冰箱或離心機中冷藏2 h,14000 r/min離心10 min,取上清液過0.22 um濾膜,上機測試。
1.2.2 標準曲線的配制
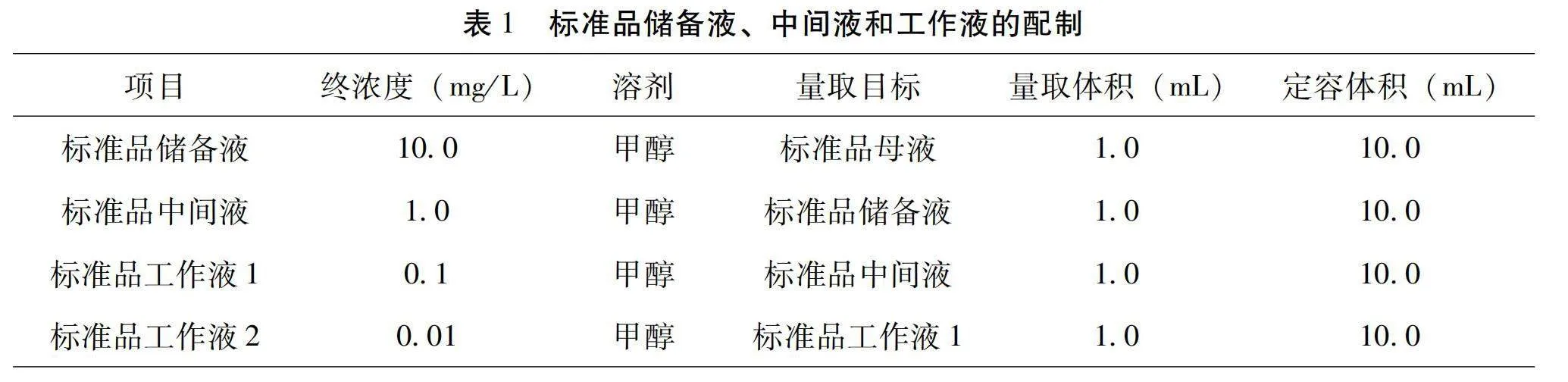
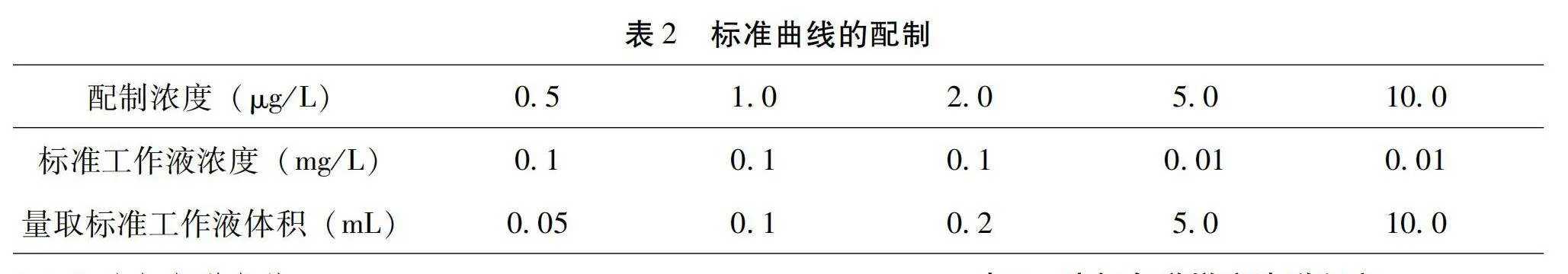
標準品母液的濃度為100 mg/L,稀釋溶劑為色譜級甲醇,量取量具為1 mL的A級移液管,定容量具為10 mL棕色容量瓶,保存溫度為-18 ℃。標準品儲備液、中間液和工作液的配制情況詳見表1,標準品工作液現用現配。
分別準確量取適量0.1 mg/L混合標準工作液1和0.01 mg/L混合標準工作液2于10 mL容量瓶中,按照1.2.1前處理流程操作,制得終濃度分別為0.5 μg/L、1.0 μg/L、2.0 μg/L、5.0 μg/L和10.0 μg/L的標準曲線,以內標法進行定量,詳見表2。
1.2.3 液相色譜條件
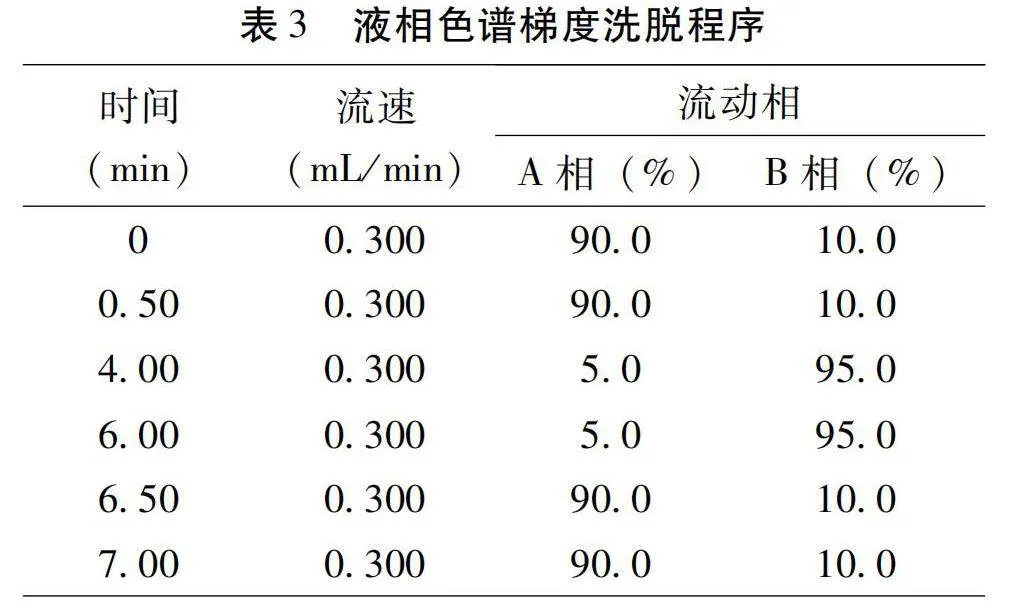
色譜柱:InfinityLab Proroshell 120 EC-C18柱(2.1 mm×50 mm×1.9 μm),流速0.300 mL/min,進樣量5 μL,柱溫30.0 ℃,流動相A:0.05%甲酸水,B:乙腈。梯度洗脫程序見表3。
1.2.4 質譜條件
配備Agilent Jet Streat的電噴霧離子源(Electrospray ionization,ESI)。目標物質在正離子條件下,多反應監測模式(DMRM)下采集參數,離子源條件、目標化合物質譜相關參數見表4、表5。
1.3 不確定度數學模型的建立
測定鯉魚中硝基呋喃類代謝物含量的數學模型為:
X=ρ×V1×V3×1000V2×m×1000(1)
式中:X為試樣中硝基呋喃類代謝物的含量(μg/kg);ρ為試樣中硝基呋喃類代謝物按內標法在標準曲線中對應的質量濃度(μg/L);V1為提取液體積(mL);V2為用于濃縮的提取液體積(mL);V3為試樣最終定容體積(mL);m為試樣稱樣質量(g)。
2 結果與分析
2.1 不確定度來源分析
根據本文實驗方法以及建立的數學模型,通過高效液相色譜串聯質譜法測定鯉魚中硝基呋喃類代謝物殘留的不確定度主要來源于:(1)隨機效應引入的不確定度urel(X);(2)稱樣量引起的不確定度urel(m);(3)標準曲線擬合引入的不確定度urel(curev);(4)標準溶液配制過程中引入的不確定度urel(c);(5)樣品定容引起的不確定度urel(V);(6)加標回收引入的不確定度urel(Rec)。
2.1.1 隨機效應引入的不確定度
在前處理包括制樣過程中的樣品均勻度、提取液和定容液的純度等一系列因素引入的不確定度,我們用平行測定6次陽性添加樣品(n=6),結果帶入貝塞爾公式進行計算得到相對標準不確定度,結果見表6。
相對標準偏差計算公式為:s(X)=∑ni=1(Xi-)2n-1(2)
標準不確定度計算公式:u(X)=s(X)n(3)
相對標準不確定度計算公式:urel(X)=u(X)(4)
2.1.2 稱量樣品引入的不確定度
本實驗室稱量樣品使用百分之一電子天平,根據檢定校準說明書最大允許誤差為±0.01 g,取矩形分布k值為3,樣品量平均為2.00 g。稱量樣品過程中引入的相對標準不確定度為:urel(m)=0.013×2.00=0.002888。
2.1.3 標準曲線擬合引入的不確定度
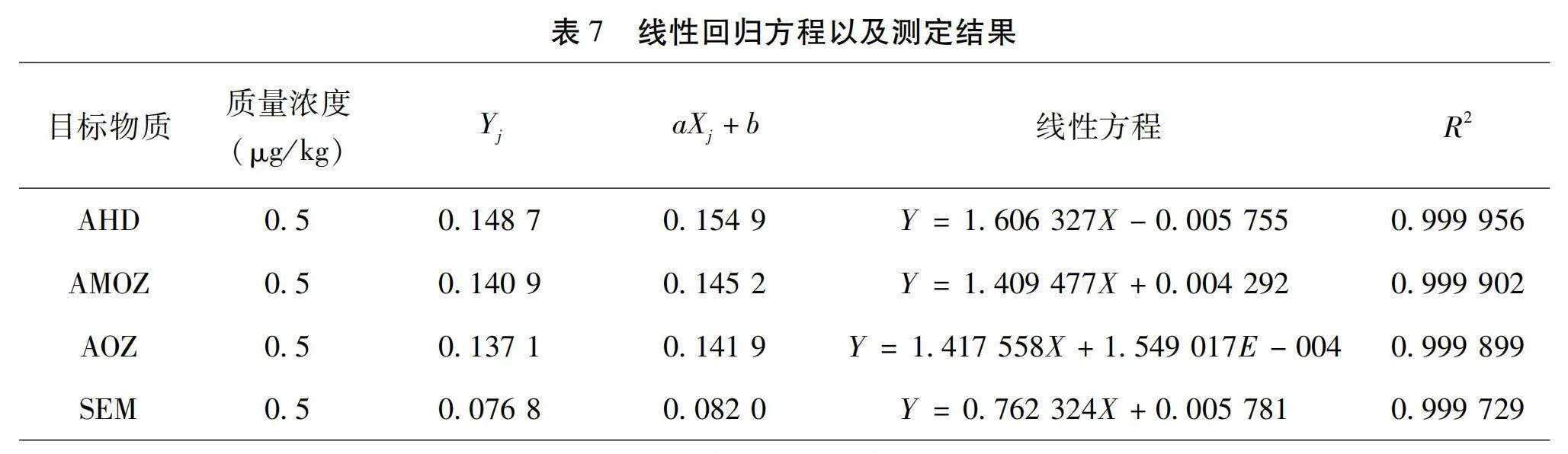
標準曲線采用最小二乘法擬合,設置5個濃度梯度,以外標響應值和內標響應值的比值Yi為縱坐標,目標化合物質量濃度為橫坐標,擬合得到各物質的線性回歸方程,格式為Y=aX+b。4種目標物的擬合曲線分別為:YAMOZ=1.409477X+0.004292、YSEM=0.762324X+0.005781、YAHD=1.606327X-0.005755、YAOZ=1.417558X+0.0001549。擬合曲線的殘余標準差能夠表示被測量估計值的分散性[8]。標準曲線擬合信息和計算結果見表7、表8。
標準曲線擬合產生的不確定度計算公式為:
u(curve)=sRa×(YQ-STD)2∑5j=1(Xj-STD)2+1n+1P(5)
殘余標準差計算公式為:
sR=∑nj=1[Yj-(b+aXj)]2n-2(6)
標準曲線擬合產生的相對標準不確定度計算公式為:
urel(curve)=u(curve)YQ(7)
式中,a為斜率,b為截距;sR為擬合曲線的殘余標準差;u(curve)為標準不確定度;Yj為第j個標準曲線濃度外標響應值比內標響應值的比值;n為標準點個數;P為樣品重復測定次數;Xj為標準曲線各點濃度;STD為標準曲線各點濃度的平均值;YQ為測定樣品平均濃度。
2.1.4 標準溶液配制過程中引入的不確定度
標準溶液配制引入的不確定度主要來源于4種硝基呋喃類代謝物及其內標物的純度、標準品定容和稀釋。
2.1.4.1 標準物質純度(P)引入的不確定度
4種硝基呋喃類代謝物及其內標物相對標準不確定度計算公式為:
u(PESTD/ISTD)=UP×3(8)
4種硝基呋喃類代謝物及其內標物合成相對標準不確定度計算公式為:
urel(Pi)=u(PESTD)2+u(PISTD)2(9)
由標準溶液使用說明書查詢相關信息帶入計算公式,取矩形分布k值為3,標準物質純度引入的不確定度結果見表9。
2.1.4.2 標準品定容引入的不確定度
標準品定容使用A級1.00 mL的移液管和10 mL的容量瓶。因此,該步驟引入的不確定度主要是定容液的膨脹因素和容量瓶的允許誤差。根據《測量不確定度評定與表示》JJF 1059.1—2012和《常用玻璃量器》JJG 196—2006要求。10 mL的容量瓶允許誤差為±0.02 mL,取矩形分布k值為3。該項引入的不確定度為:u(Vp1)=0.023=0.01155。
本實驗條件下實驗室溫度在18~23 ℃浮動,定容液甲醇在該溫度范圍體積膨脹系數為1.2×10-3/℃,均勻分布下k值為3,由膨脹因素引入的不確定度為:
u(Vp2)=1.2×10-3×5×103=0.03464。
綜上所述,容量瓶引入的相對合成不確定度為:
urel(STD1)=u(Vp1)2+u(VP2)210=0.001275。
A級1.00 mL的移液管允許誤差為±0.008 mL,在上述實驗溫度下,定容液為甲醇,考慮體積膨脹系數,取均勻分布k值為3,標準不確定度為:
u(VP3)=0.0083=0.004619
u(VP4)=1.00×5×1.2×10-33=0.003464。
移液管相對合成標準不確定度為:
urel(STD2)=u(VP3)2+u(VP4)21.00=0.005774。
綜上所述,標準物質定容引入的相對合成標準不確定度為:
urel(Sv)=urel(STD1)2+urel(STD2)2=0.01170。
2.1.4.3 標準品稀釋引入的不確定度
實驗使用的標準工作液為外標1000 μg/L和100 μg/L,內標1000 μg/L,稀釋過程中使用到A級1.00 mL的移液管3次,10 mL的容量瓶3次。根據(2)中所述條件,由標準物質稀釋引入的相對合成標準不確定度為:
urel(Sd)=urel(STD1)2×3+urel(STD2)2×3=0.003925。
綜上所述,標準溶液配制過程引入的標準不確定度為:
urel(c)=urel(Pi)2+urel(Sv)2+urel(Sd)2。
2.1.5 樣品定容引起的不確定度
樣品定容使用A級1.00 mL的移液管,允許誤差為±0.008 mL,樣品定容引入的不確定度為:
u(VP3)=0.0083=0.004619。
定容液為95%的甲醇水,樣品定容不確定度來源于溫度變化時液體體積的變化,因此僅考慮液體變化情況,用純水膨脹系數近似代替定容液膨脹系數,為2.08×10-4/℃,實驗室溫度變化范圍±5 ℃,取矩形分布k值為3,由膨脹因素引入的不確定度為:
u(Vp5)=1.00×5×2.08×10-43=0.0006004。
因此,樣品定容相對合成不確定度為:
urel(V)=u(VP3)2+u(VP5)21.00=0.000622。
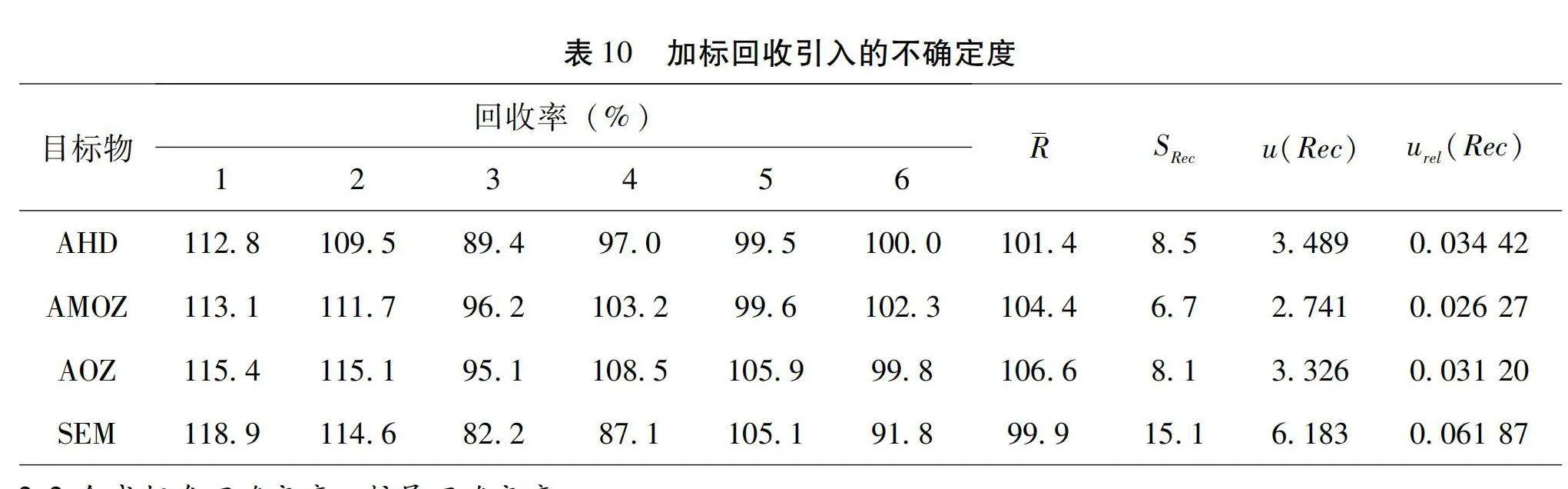
2.1.6 加標回收引入的相對不確定度
實驗中添加濃度為5.00 μg/kg的混合標準溶液,平行測定6次,加標回收相對不確定度根據貝塞爾公式計算結果見表10。公式為:
u(Rec)=SRec6(10)
urel(Rec)=u(Rec)(11)
公式中urel(Rec)為加標回收引入的相對標準不確定度,為回收率平均值,SRec為回收率標準偏差,u(Rec)為加標回收引入的標準不確定度。
2.2 合成標準不確定度、擴展不確定度
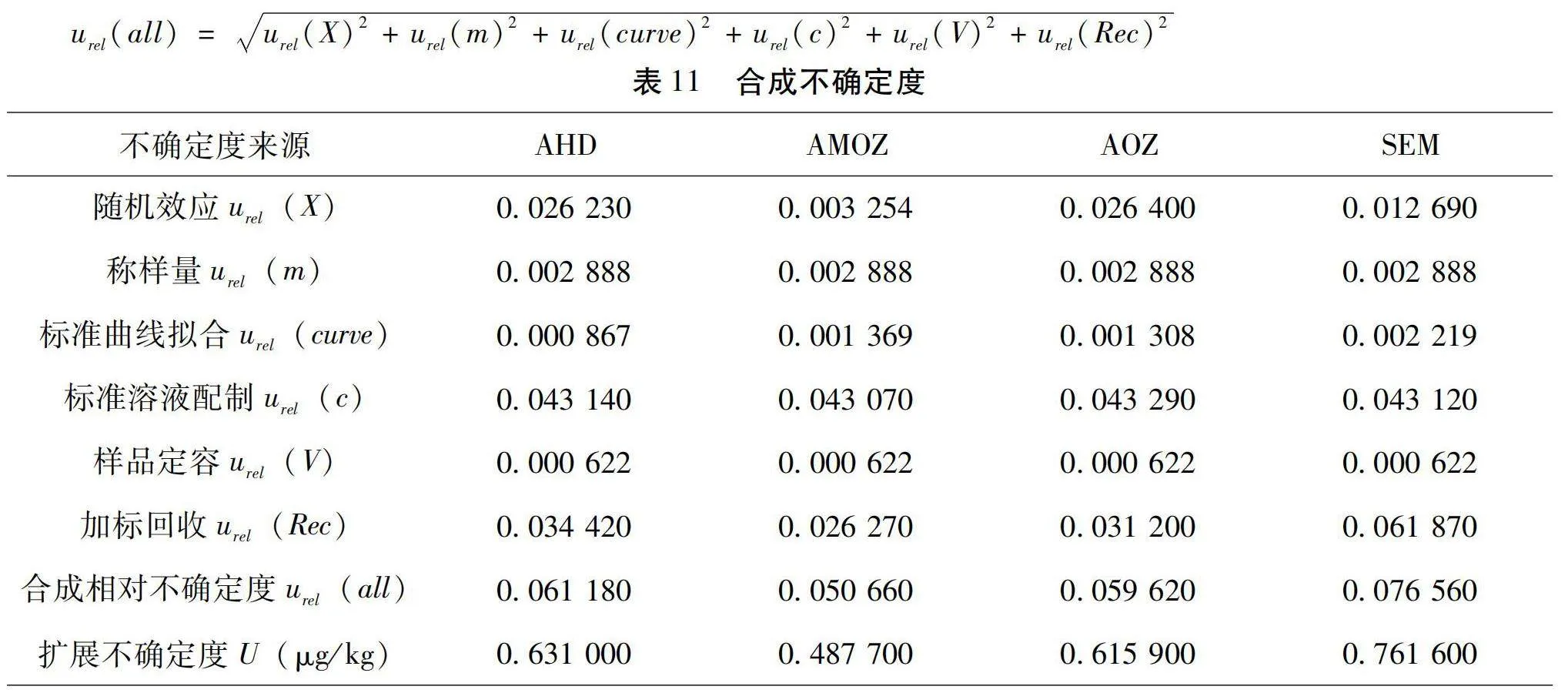
全過程合成不確定度計算公式如下,計算結果見表11。
urel(all)=urel(X)2+urel(m)2+urel(curve)2+urel(c)2+urel(V)2+urel(Rec)2
在置信率為95%時,包含因子K=2,樣品檢測結果平均值,擴展不確定度為:
U=urel(all)×K×
使用本方法檢測鯉魚中4種硝基呋喃代謝物檢測結果最終報出為:AHD=5.157±0.631 0 μg/kg,AMOZ=4.813±0.487 7 μg/kg,AOZ=5.165±0.615 9 μg/kg,SEM=4.974±0.761 6 μg/kg。
3 討論與結論
本研究表明,利用高效液相色譜-串聯質譜法測定鯉魚中硝基呋喃類代謝物殘留量的實驗過程中,隨機效應、標準溶液配制和加標回收為本實驗不確定度的主要來源,這一結果與楊魯群等[9]、林功師[10]、張惠峰等[11]的評估結果相似。而在標準溶液配制過程中標準品純度和定容所使用的量器引入的不確定度,遠高于標準品稀釋。樣品定容、標準曲線擬合以及稱樣量所帶來的不確定度較小。因此,在日常檢測中可以通過購買純度更高的標準品、采購精度更高的玻璃量器或者移液器以及避免過多的逐級稀釋母液的方式來降低檢測過程中的不確定度,來保證檢測結果的準確可靠。
參考文獻
[1]呂燕.水產品中硝基呋喃類藥物代謝殘留的不確定度評定[J].浙江農業科學,2022,63(9):2098-2102+2106.
[2]McCalla D R.Mutagenicity of nitrofuran derivatives:review[J].Environmental Mutagenesis,1983,5(5):745-765.
[3]McCracken R J,Kennedy D G.Determination of the furazolidone metabolite,3-amino-2-oxazolidinone,in porcine tissues using liquid chromatography-thermospray mass spectrometry and the occurrence of residues in pigs produced in Northern Ireland[J].Journal of Chromatography B:Biomedical Sciences and Applications,1997,691(1):87-94.
[4]譚志軍,翟毓秀,冷凱良,等.呋喃西林和呋喃唑酮代謝物在大菱鲆組織中的消除規律[J].中山大學學報(自然科學版),2008,47(S1):63-69.
[5]趙東豪,黎智廣,李劉冬,等.蝦苗使用呋喃西林和呋喃唑酮的殘留評估[J].南方水產科學,2012,8(3):54-58.
[6]魏涯,岑劍偉,郝淑賢,等.高效液相色譜法測定池塘底泥中呋喃西林的研究[J].南方水產科學,2014,10(1):71-77.
[7]唐紅梅,曾芳,李成洪.食品中硝基呋喃類藥物及其代謝物殘留檢測的研究進展[J].食品安全質量檢測學報,2016,7(10):3952-3959.
[8]弓浩然,洪冰,胡婷婷,等.ICP-MS測定小麥中5種金屬元素的不確定度評價[J].糧食科技與經濟,2021,46(6):83-88.
[9]楊魯瓊,裴華,朱曉薇,等.液相色譜-串聯質譜法測定魚餌中地西泮的不確定度評定[J].飼料研究,2024,47(2):122-126.
[10]林功師.UHPLC-MS/MS測定羅非魚中硝基呋喃類代謝物殘留量的不確定度評定[J].中國漁業質量與標準,2017,7(6):48-57.
[11]張惠峰,綦天華.高效液相色譜法測定水產品中甲基睪酮殘留量的不確定度評定[J].食品科學,2015,36(18):199-203.
猜你喜歡
分析化學(2017年1期)2017-02-06 21:32:17
中國醫藥導報(2016年30期)2016-12-28 12:18:02
熱帶農業科學(2016年10期)2016-12-12 01:52:56
分析化學(2016年7期)2016-12-08 00:57:07
上海醫藥(2016年21期)2016-11-21 23:14:07
中國科技博覽(2016年18期)2016-10-19 11:09:28
科學與財富(2016年28期)2016-10-14 04:01:52
中國科技博覽(2016年2期)2016-04-25 14:06:58
上海醫藥(2016年3期)2016-03-23 23:38:20
現代儀器與醫療(2015年4期)2015-07-15 10:13:19
