ERK5抑制劑XMD17-109通過下調ITPRIP表達抑制膠質瘤進展*
2023-12-26 02:25:52王昕雯曹長春孫增先
重慶醫學 2023年23期
關鍵詞:實驗
王昕雯,曹長春,朱 亮,杜 宇,孫增先
(1.徐州醫科大學附屬連云港醫院藥學部,江蘇連云港 222061;2.徐州醫科大學淮安臨床學院藥學部,江蘇淮安 223300;3.南京醫科大學附屬淮安第一醫院藥學部,江蘇淮安 223300)
膠質瘤(glioblastoma,GBM)被認為是惡性程度最高、治療難度最大的原發性腦腫瘤之一,發病率占惡性腦腫瘤的45%以上[1-3]。盡管多學科管理和靶向治療在臨床方面取得了一定進展,但惡性膠質瘤的現有治療方案仍然較為局限[4-5]。抑癌基因的失活是膠質瘤不斷進展的重要原因,也是治療的主要障礙之一[6-8]。因此,深入了解這一過程所涉及的分子機制對于開發更有效的干預措施至關重要。細胞外信號調節激酶5(extracellular signal-regulated kinase 5,ERK5)是絲裂原活化蛋白激酶家族中的一員,具有獨特的C末端結構域[9-10]。越來越多的證據表明ERK5參與腫瘤起始和進展的各個階段[11-13]。研究認為,ERK5信號級聯的激活會誘導多種細胞周期分子增殖[14]。此外,ERK5對周期蛋白依賴性激酶抑制因子的表達具有下調作用[15]。藥物干預和基因策略明確證實,靶向ERK5通路對于癌癥治療具有潛力[16-17]。基因表達譜分析發現ERK5 mRNA在原發性腦腫瘤,特別是GBM中明顯高表達[18]。新型小分子激酶抑制劑XMD17-109作為ERK5強效抑制劑被成功合成。XMD17-109在生化上抑制ERK5,IC50為(0.162±0.006)μmol/L,在細胞內阻斷表皮生長因子誘導的ERK5自磷酸化。目前XMD17-109用于膠質瘤治療的研究尚未見報道,且XMD17-109作用機制有待進一步探究。1,4,5-三磷酸受體(inositol 1,4,5-trisphosphate receptor,IP3R)相互作用蛋白已被證明是IP3R的結合伴侶,其與IP3R相互作用促進IP3R通道的鈣離子抑制功能。前期研究已經證明肌醇1,4,5-三磷酸受體相互作用蛋白(inositol 1,4,5-trisphosphate receptor interacting protein,ITPRIP)通過連接肌球蛋白調節輕多肽9和死亡相關蛋白激酶1促進GBM進展[19]。本研究假設ERK5在膠質瘤中可能作為腫瘤促進ITPRIP的刺激分子。通過研究ERK5抑制劑XMD17-109對膠質瘤細胞U251活力的影響及ERK5與ITPRIP之間的相互作用,可揭示ERK5介導促進ITPRIP表達的分子機制,及其在膠質瘤進展中的意義,現報道如下。
1 材料與方法
1.1 材料
1.1.1細胞
本研究中人膠質母細胞瘤細胞株U251來自中國科學院分子細胞科學卓越創新中心。
1.1.2試劑
XMD17-109購于美國MCE公司;DMEM高糖培養基和胎牛血清(FBS)購于美國Gibco公司;Lipofectamine 2000轉染試劑購于美國Thermo Fisher公司;PrimeScriptTMRT Master Mix和SYBR &Premix Ex TaqTM試劑盒購于日本TaKaRa公司;BCA試劑盒和十二烷基硫酸鈉-聚丙烯酰胺凝膠電泳(sodium dodecyl sulfate-polyacrylamide gel electrophoresis,SDS-PAGE)試劑購于上海碧云天生物技術有限公司;ERK5抗體購于美國Cell Signaling公司(1∶1 000,#12950);ITPRIP抗體(1∶1 000,26055-1-AP)和GAPDH抗體(1∶5 000,60004-1-Ig)購于美國Proteintech公司;辣根過氧化物酶(HRP)-山羊抗小鼠IgG抗體(1∶5 000,PA2201)和HRP-山羊抗兔IgG抗體(1∶5 000,PA2202)購于南京普諾恩生物技術有限公司;CCK8檢測試劑盒購于南京建成公司;7-氨基放線菌素D(7-AAD)/藻紅蛋白(PE)試劑盒和Transwell小室購于美國BD公司;ERK5、陰性對照siRNA(siNC)序列和質粒由上海GenePharma公司設計并合成。
1.1.3儀器
Applied biosystemsTM7500熒光定量PCR儀、酶標儀、培養箱和超凈臺購于美國Thermo Fisher公司;流式細胞儀購于美國Beckman公司;高速冷凍離心機購于芬蘭Eppendorf公司;Amersham Biosciences蛋白成像系統購于美國Cytiva公司。
1.2 方法
1.2.1細胞培養
細胞在含有10% FBS的DMEM高糖培養基中,37 ℃、5% CO2環境常規培養。在體外細胞實驗中,使用濃度為1 μmol/L的XMD17-109處理細胞24 h以抑制ERK5活性(XMD17-109組),未處理的細胞納入Control組。
1.2.2RNA干擾
按照說明書提示方法,分別將ERK5 siRNA(siERK5)和siNC使用Lipofectamine 2000進行細胞轉染,構建ERK5敲低和陰性對照模型,分為siERK5組、siNC組。轉染6 h后,更換為含10% FBS的新鮮培養基。細胞連續培養48 h后,收集細胞進行蛋白、RNA分離等研究。通過逆轉錄實時熒光定量PCR(RT-qPCR)測定ERK5敲低效率。siERK5組和siNC組所對應RNA序列分別為5′-CGU GCC CUA UGG CGA AUU CAA TT-3′和5′-UUC UCC GAA CGU GUC ACG UTT-3′。
1.2.3質粒轉染
人ERK5基因經過擴增和逆轉錄合成互補DNA(cDNA)。隨后,將人ERK5 cDNA克隆pCDH-CMV-MCS-EF1-copGFP-T2A-Puro載體中。將含有ERK5和對照結構的溶液引入到U251細胞中,孵育48 h,構建ERK5過表達和對照結構模型,分別分為ERK5-OE組、Vector組。采用RT-qPCR和Western blot檢測證實ERK5在細胞中的過表達水平。進一步將ERK5-OE組、Vector組細胞分別與XMD17-109進行孵育,分為ERK5-OE+XMD17-109組、Vector+XMD17-109組。
1.2.4總RNA提取及RT-qPCR
轉染siRNA后,用TRIzol試劑提取細胞總RNA。使用納米微滴設備評估總RNA的純度和濃度。使用PrimeScriptTMRT Master Mix進行RNA逆轉錄,然后通過SYBR &Premix Ex TaqTM進行RT-qPCR檢測。采用Ct(2-ΔΔCt)法(ΔΔCt=ΔCttarget-ΔCtGAPDH)對所有靶基因進行標準化,以GAPDH為內參基因。引物序列見表1。

表1 RT-qPCR引物序列
1.2.5Western blot
細胞收集后在冰上冷卻30 min,使用含有20 mmol/L Tris、1 mmol/L EDT A、150 mmol/L氯化鈉、1 mmol/L乙二醇雙(2-氨基乙基醚)四乙酸(EGTA)、1%脫氧膽酸鹽、1% Triton X-100,2 mmol/L正釩酸鈉的裂解緩沖液進行蛋白質提取。隨后,裂解液在4 ℃下以13 000 r/min離心20 min。所得上清液使用BCA試劑盒測定。將每個樣品中等量的蛋白質與SDS樣品緩沖液相結合,通過SDS-PAGE分離蛋白質,并將其轉移到聚偏二氟乙烯(PVDF)膜上。蛋白轉移完畢后,用含有5%脫脂奶粉的TBST(TBS中含0.1%吐溫20)封閉1 h。然后將膜與ERK5抗體、ITPRIP抗體和GAPDH抗體在4 ℃下過夜孵育。用TBST清洗3次,每次清洗5 min。加入二抗,室溫下孵育1 h,其中ERK5和ITPRIP選用HRP-山羊抗兔IgG二抗,GAPDH選用HRP-山羊抗小鼠IgG二抗。用增強化學發光檢測系統對膜進行檢測。使用Image J軟件進行吸光度進行分析,每個蛋白條帶的信號強度基于相對于各自的GAPDH加載對照進行歸一化。
1.2.6CCK-8和細胞增殖實驗
使用CCK-8檢測試劑盒評估細胞增殖能力。在96孔板中每孔加入100 μL密度為3×104/mL的細胞混懸液。在5% CO2的培養箱中孵育0、24、48、72 h。隨后,每孔加入10 μL的CCK-8試劑,繼續孵育4 h后用酶標儀測定450 nm處吸光度(A)值,每組條件均設置3個復孔。
1.2.7流式細胞儀檢測細胞凋亡
采用7-AAD/PE法檢測U251細胞凋亡情況。將U251細胞與7-AAD/PE在冰上孵育15 min,檢測細胞凋亡情況,應用Flowjo軟件進行定量分析。
1.2.8劃痕實驗和Transwell實驗
在劃痕實驗中,將細胞以2×106個/孔的密度重懸于6孔板中。一旦細胞貼壁,使用20 μL移液器吸頭在細胞表面做出劃痕。每個實驗組均設置3個復孔。分別于劃痕后0、24 h在顯微鏡下觀察并拍照。每個孔中測量3個隨機位置,使用Image J軟件量化劃痕寬度。按照說明書提示方法使用Transwell小室進行細胞遷移和侵襲實驗。將1×105個細胞接種于上室500 μL無血清培養基中,下室加入750 μL含10% FBS的新鮮培養基,將細胞在37 ℃下孵育36 h。然后將遷移過濾網的細胞用4%多聚甲醛(PFA)固定,并用結晶紫溶液染色20 min。用棉簽去除殘留在上室表面的細胞后,在顯微鏡下隨機拍照5個視野,采用直接計數法用Image J軟件對遷移和侵襲細胞數量進行計數,取平均值作為最終遷移或侵襲細胞數量。
1.3 統計學處理
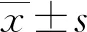
2 結 果
2.1 XMD17-109對ERK5和ITPRIP的影響
Control組與XMD17-109組ERK5 mRNA和蛋白表達水平無明顯變化;與Control組相比,XMD17-109組ITPRIP mRNA和蛋白表達水平明顯降低(P<0.05);與Control組相比,XMD17-109組吸光度降低,U251細胞增殖、侵襲和遷移能力均明顯降低,細胞凋亡率明顯升高,差異有統計學意義(P<0.05),見圖1、2。

A:ERK5 mRNA表達水平;B:細胞增殖能力;C:劃痕實驗結果(100×);D:遷移實驗結果(200×);E:侵襲實驗結果(200×);F:流式細胞術分析結果;①:Control組;②:XMD17-109組;ns:P>0.05,與Control組比較;a:P<0.05,與Control組比較。
2.2 敲低與過表達ERK5對U251細胞的影響
與siNC組比較,siERK5組ERK5和ITPRIP mRNA和蛋白表達水平明顯下調;與Vector組比較,ERK5-OE組ERK5和ITPRIP mRNA和蛋白表達水平明顯上調;與siNC組比較,siERK5組U251細胞增殖、侵襲和遷移能力均明顯降低,細胞凋亡率明顯升高(P<0.05);與Vector組比較,ERK5-OE組U251細胞增殖、侵襲和遷移能力均明顯升高(P<0.05),見圖2~4。

A:8組ITPRIP mRNA表達水平;B:8組ERK5和ITPRIP蛋白Western blot結果;C:8組ERK5蛋白表達水平;D:8組ITPRIP蛋白表達水平;①Control組;②XMD17-109組;③siNC組;④siERK5組;⑤Vector組;⑥ERK5-OE組;⑦Vector+XMD17-109組⑧ERK5-OE+XMD17-109組;a:P<0.05,與Control組比較;b:P<0.05,與siNC組比較;c:P<0.05,與Vector組比較;d:P<0.05,與Vector+XMD17-109組比較。

A:ERK5 mRNA表達水平;B:細胞增殖能力;C:劃痕實驗結果(100×);D:遷移實驗結果(200×);E:侵襲實驗結果(200×);F:流式細胞術分析結果;①:SiNC組;②:SiERK5組;a:P<0.05,與SiNC組比較。
2.3 XMD17-109逆轉ERK5對U251細胞的調控作用
與Vector+XMD17-109組比較,ERK5-OE+XMD17-109組ERK5和ITPRIP mRNA和蛋白表達水平明顯上調;與Vector+XMD17-109組比較,ERK5-OE+XMD17-109組U251細胞增殖、侵襲和遷移能力均明顯升高,細胞凋亡率明顯降低,見圖2、5。
3 討 論
原發或復發的膠質瘤起源于分化細胞,如成熟的神經元、星形膠質細胞和成體神經干細胞[20-21]。這一現象表明,惡性膠質瘤可能保留神經生物學特征和信號,使得神經中心膠質瘤治療成為一種有前途的方法[22-24]。ERK5在調節多種凋亡信號通路中發揮重要作用,不僅促進腫瘤生長,還參與腫瘤細胞的增殖[25-26]。另一方面,ITPRIP缺陷已被證明會加重短暫性腦缺血和急性興奮性毒性后的神經毒性[27]。基于這一認識,作者認為在膠質瘤細胞中,ERK5可能通過蛋白-蛋白相互作用對腫瘤啟動子ITPRIP發揮促進作用,類似于其在神經元細胞中的作用。
ERK5在癌癥中的致癌作用歸因于其靶向特定的mRNA[28]。采用siRNA/shRNA沉默或敲除ERK5基因的多項研究表明,ERK5信號通路對多種細胞表型發揮重要調控作用。研究數據顯示,體內和體外實驗中沉默ERK5均能抑制多種癌細胞細胞的增殖作用,如子宮內膜癌、非小細胞肺癌、小細胞肺癌、黑色素瘤、三陰性乳腺癌和膽管癌等[29]。在小細胞肺癌中,沉默ERK5后細胞相較于野生型細胞增殖活力明顯降低,這一結果強調了ERK5激酶活性在小細胞肺癌細胞增殖中的重要作用。此外,一項針對實驗用增殖表皮癌細胞的研究顯示,表皮生長因子能夠引起ERK5的活化[30]。ERK5通過調節基質相關基因、促血管生成因子和整合素維持三陰性乳腺癌細胞的遷移和間質表型[17,31]。一項關于骨肉瘤的研究顯示,ERK5是一種有效的預后指標,調節ERK5-STAT3通路會加劇骨肉瘤細胞的惡性表型[32]。
通過數據庫對腫瘤基因表達譜進行分析,結果表明ERK5與ITPRIP在膠質瘤組織中的表達上調。基于此,作者推測ITPRIP在膠質瘤中的作用可能是通過調控ERK5軸介導。為證實這一假設,本研究進行了多項功能實驗,發現XMD17-109處理后細胞表型明顯抑制,ERK5表達無明顯變化,ITPRIP表達降低。這是因為XMD17-109作為ERK5選擇性抑制劑,通過阻斷細胞內表皮生長因子誘導的ERK5自磷酸化,對ERK5下游分子發揮抑制作用,但其對ERK5本身無明顯抑制作用。敲低ERK5會抑制膠質瘤細胞增殖、遷移和侵襲能力,誘導細胞凋亡,下調ITPRIP表達。相反,過表達ERK5對膠質瘤細胞的生物學功能起到促進作用,同時上調ITPRIP表達。ERK5抑制劑XMD17-109減弱了膠質瘤細胞增殖、遷移和侵襲能力,同時能夠逆轉過表達ERK5對膠質瘤細胞表型的促進作用。此外,前期研究證實,敲低ITPRIP對于膠質瘤細胞表型具有抑制作用[19],因此可以推論:XMD17-109通過阻斷ERK5自磷酸化抑制ITPRIP表達,進而抑制膠質瘤細胞表型。
綜上所述,本研究證實了XMD17-109通過抑制ERK5/ITPRIP信號通路進而抑制膠質瘤的進展,ERK5、ITPRIP可以作為膠質瘤預防、診斷和治療的潛在生物標志物。
猜你喜歡
作文·小學低年級(2025年2期)2025-02-13 00:00:00
小雪花·小學生快樂作文(2024年11期)2024-12-31 00:00:00
作文·小學低年級(2024年2期)2024-04-29 00:00:00
作文·小學低年級(2023年3期)2023-04-29 00:00:00
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
小主人報(2022年4期)2022-08-09 08:52:06
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
