MYH7基因變異相關兒童心肌病表型與基因型特點的回顧性分析
2023-12-06 08:29:34劉露鄭奎張英謙
中國當代兒科雜志 2023年11期
劉露 鄭奎 張英謙
(1.河北北方學院研究生學院,河北張家口 075000;2.河北省兒童醫院心內科/河北省小兒心血管重點實驗室,河北石家莊 050031;3.重慶醫科大學附屬璧山醫院,重慶 402760)
心肌病(cardiomyopathy, CM)是以心肌結構和功能異常為特征,無法用負荷異常、冠狀動脈疾病或先天性缺陷來解釋的心肌疾病[1]。CM在兒童中罕見,常導致心源性猝死(sudden cardiac death, SCD)或心力衰竭(heart failure, HF),病死率高[2]。擴張型心肌病(dilated cardiomyopathy,DCM) 占兒童CM 的50%~60%,肥厚型心肌病(hypertrophic cardiomyopathy, HCM) 占30%~40%,致密化不全型心肌病(left ventricular noncompaction cardiomyopathy, LVNC)約占5%[3]。兒童CM的發病機制及病因復雜,不同病因預后差異大,其中遺傳病因占有重要地位。目前發現40 余種基因與兒童CM 相關,常見的致病基因包括MYH7、TTN、LMNA等基因[4]。其中MYH7基因是兒童HCM最常見的致病基因[5]。CM臨床異質性強,同一基因變異可導致不同臨床表型,且與患兒預后顯著相關[6]。當前國內對兒童CM的遺傳研究報道較少。本研究通過分析5例MYH7基因變異所致的CM 患兒的臨床資料,總結其基因型-臨床表型及隨訪情況,為精準評估MYH7基因變異相關CM患兒預后提供參考。
1 資料與方法
1.1 研究對象
回顧性收集2021 年4 月—2022 年11 月于河北省兒童醫院心內科確診的5例MYH7基因變異所致CM 患兒的臨床資料。CM 診斷標準參考《AHA 兒童心肌病的分類和診斷科學聲明解讀》[7]。
本研究通過河北省兒童醫院醫學倫理委員會批準(202222-67),患兒監護人簽署知情同意書。
1.2 資料采集
通過查閱電子病歷系統收集患兒資料,包括首診年齡、發育情況、起病特征、超聲心動圖、心電圖(electrocardiogram, ECG)、心肌酶、自身抗體譜、血尿遺傳代謝篩查等結果。通過門診或電話獲得隨訪資料,包括超聲心動圖及臨床結局等。隨訪時間截至2023年5月1日。
采用全基因組測序、全外顯子組測序、Panel技術對患兒進行基因檢測,測序結果經Sanger測序驗證變異來源。最后根據美國醫學遺傳學和基因組學學會(American College of Medical Genetics and Genomics,ACMG)相關指南[8]和ClinGen 專家共識(https://www.clinicalgenome.org/)對變異的致病性進行評估。
2 結果
2.1 一般資料及臨床表現
5例CM患兒中,女3例,男2例,中位發病年齡為36(范圍:6~156)個月。首診時臨床表現為咳嗽、呼吸急促、吃奶欠佳、口唇發紺(HF表現)2 例,均生長發育落后,其中1 例伴雙下肢水腫和肌張力減低;心悸、黑蒙、暈厥1例;發熱、流涕伴肝功能異常1 例。5 例均存在活動耐力減低、乏力。4 例有心功能不全(改良ROSS 評分),2 例有CM家族史。見表1。

表1 5例CM患兒的臨床資料
2.2 首診超聲心動圖、ECG和實驗室檢查結果
5例CM患兒首診超聲心動圖及ECG均有異常。LVNC(病例1)和DCM(病例3)患兒超聲心動圖提示心室擴大,心肌組織疏松、增厚,肌小梁粗大,左室收縮功能減低且伴瓣膜反流,左室射血分數(left ventricular ejection fraction, LVEF)和左室短軸縮短率低于正常值(LVEF 正常值:>55%;左室短軸縮短率正常值:>25%。)。3 例(病例2、4、5)HCM 患兒室間隔厚度均高于正常值且左室舒張功能減低,其中2例(病例4、5)存在心尖部增厚伴二、三尖瓣輕度反流,1 例(病例5)有少量心包積液。5例ECG均有ST-T段改變,4例(病例1、2、4、5)左室高電壓,3例(病例1、3、4)竇性心動過速及1 例(病例2)竇性心動過緩伴不齊,2例(病例3、4)P波高尖,1例(病例3)PR間期延長。
5 例CM 患兒心肌酶譜均異常升高,以α-羥丁酸脫氫酶為主(5/5),其次為乳酸脫氫酶(4/5)、谷草轉氨酶(2/5)、肌酸激酶同工酶(1/5)。5 例腦鈉肽均高于正常值,LVNC和DCM患兒升高更顯著。病例1肌鈣蛋白Ⅰ升高。病例3自身抗體譜中抗ds-DNA 弱陽性。5 例患兒尿有機酸檢測未見異常;2例(病例3、5)血氨基酸和酰基肉堿檢測異常,病例3多種肉堿升高,病例5瓜氨酸升高。
2.3 基因檢測結果
5 例CM 患兒均為MYH7基因(NM_000257)雜合變異,其中2例為復合雜合變異,共包括7種不同的變異位點(5種為新發現變異),5種錯義變異,1 種整碼缺失變異,1 種剪接變異,具體變異位點及致病性分析見表2。
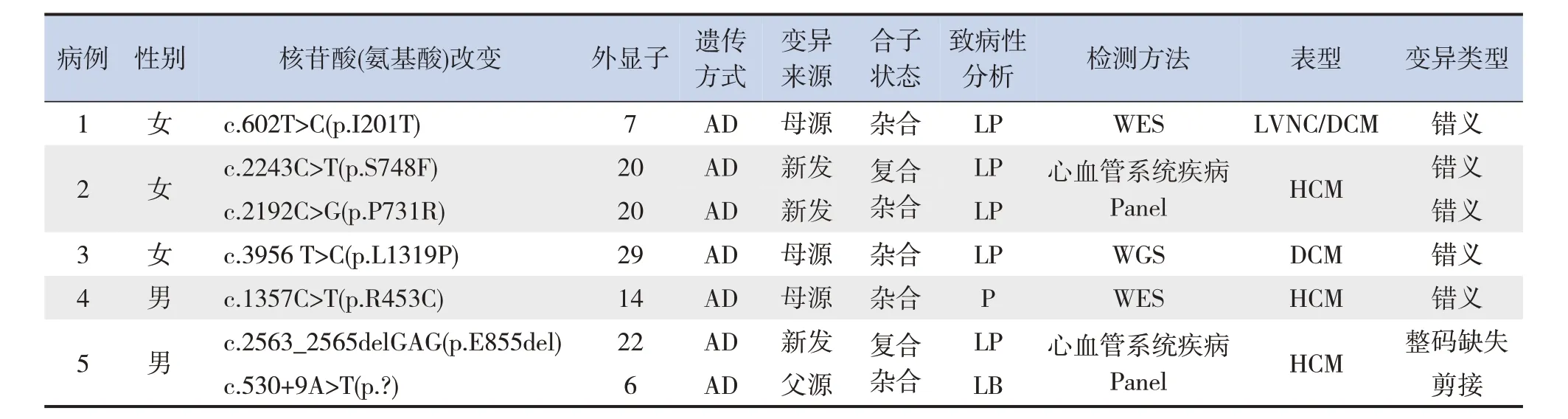
表2 5例CM患兒的基因檢測結果
2.4 治療及隨訪
5 例CM 患兒末次隨訪中位年齡為60(范圍:12~170)個月,經改善心功能藥物治療后,5例患兒臨床表現均好轉。LVNC(病例1)及DCM(病例3) 患兒左室收縮功能恢復,LVEF (64%、70%)和左室短軸縮短率(34%、38%)良好。3例HCM 患兒心室舒張功能稍減低,2 例(病例4、5)室間隔厚度改善,但二、三尖瓣反流和少量心包積液仍存在。中位隨訪時間為16(范圍:7~24)個月,5 例患兒的心肌肥大及纖維化未明顯逆轉,但均存活。
3 討論
MYH7基因變異是CM常見的遺傳學病因之一。30%~50%的兒童HCM 與MYH7基因變異有關,1%~5.3%的MYH7基因變異可導致DCM,成人LVNC 患者的MYH7基因變異約占48%[5]。MYH7基因變異位置、變異類型及遺傳模式與臨床表型和起病時間密切關聯[10]。MYH7基因頭部(S1)和頸部變異率最高,多與HCM 表型相關。因為S1存在多個關鍵功能區域,可能影響心肌能量供應。α 螺旋尾部變異多見于DCM 表型,與兒童早期發病有關[11-12]。MYH7基因致病變異多為常染色顯性遺傳的錯義變異,主要通過改變編碼氨基酸的組成影響蛋白質的結構和功能[13]。兒童CM具有顯著的遺傳和臨床表型異質性,致病變異與患兒預后不良密切相關[14]。
MYH7基因變異所致兒童HCM 在疾病早期缺乏特異性表現,50%~60%患兒青春期出現肌節蛋白遺傳缺陷相關臨床表現[15],多以運動時呼吸困難、胸痛、心悸、暈厥或SCD為主[16]。本研究中3例HCM 患兒發病年齡(均≥3 歲)晚于DCM 及LVNC 患兒(均<1 歲),2 例以活動耐力差表現為主,1例年長兒(13歲)表現為活動后心悸、意識喪失。有研究報道,盡管MYH7基因變異所致HCM 發病相對較晚,但具有潛在HF 和SCD 風險;兒童時期發病的HCM 常與復雜臨床表型和主要心血管不良事件(major adverse cardiovascular event,MACE)相關[17]。Tajsharghi 等[18]報道1例10 月齡確診MYH7基因變異的HCM 患兒,于11 歲時猝死。因此,HCM 一旦明確診斷,不論臨床表現是否典型,都應密切隨診,警惕SCD 和MACE 發生。多數HCM 患者有ECG 異常,2%~5%的HCM 患者可伴預激綜合征或房室結折返性室上性心動過速[11]。本研究中3例HCM患兒ECG以ST-T段改變和左室高電壓為主,其中1 例(病例2)有竇性心動過緩伴不齊表現,1 例(病例4)存在竇性心動過速伴P 波高尖。60% HCM 患者有家族史,Marschall等[19]報道1例MYH7基因c.2192C>T變異的HCM 患者于18 歲時猝死,其母親是DCM 患者。本研究中3例HCM患兒雖無家族史,但病例2與上述報道的變異位點相同而變異堿基不同(c.2192C>G),需警惕患兒出現SCD,應定期隨訪。MYH7基因存在“基因劑量效應”,復合雜合變異可能導致更嚴重的臨床表型[12]。本研究中2例(病例2、5)HCM 患兒存在復合雜合變異,臨床表型較病例4嚴重,與上述報道[12]一致。病例5 患兒的2 個變異中c.530+9A>T 變異評級為可能良性,此變異是父源,而父親目前無表型,提示患兒HCM 表型可能僅由c.2563_2565delGAG變異控制。
1%~5%的DCM 是由MYH7基因變異所致[15]。兒童DCM 發病年齡早,多見于嬰幼兒,臨床表現更嚴重,常以HF 為主要表現,20%病例有家族史[10,13,20]。本研究病例3首診年齡為6個月,以反復咳嗽、呼吸急促、哭鬧時口唇發紺、雙下肢水腫等嚴重HF表現為主,另有吃奶欠佳、乏力、發育落后及肌張力減低等臨床特點,母親有先天性心臟病史。該患兒存在MYH7基因L1319P 變異,位于α 螺旋尾部。Armel 等[21]研究表明,位于MYH7基因α 螺旋尾部變異導致的DCM 表型嚴重,可能是變異影響肌絲組裝導致熱力學穩定性下降而改變粗絲的正確裝配。Meredith等[22]報道MYH7基因R1500P 位點變異破壞肌球蛋白尾部形成卷曲螺旋的能力導致Laing 早發型遠端肌病。本研究中病例3 的L1319P 變異也位于MYH7基因α 螺旋尾部,故在該患兒的長期隨訪過程中,還應關注是否出現Laing遠端肌病表型。
有研究顯示,MYH7基因變異是發生LVNC 的獨立危險因素[23]。孤立性LVNC 罕見,常合并其他表型,以DCM 表型最常見。本研究病例1 主要診斷為LVNC,但同時合并DCM表型。MYH7基因變異所致LVNC患兒發病早,多在<1歲時確診,常表現為臨床三聯征:HF、室性心律失常和血栓栓塞[24]。病例1 首診年齡6 月齡,以咳嗽為主要表現,伴有吃奶欠佳、呼吸急促、口唇發紺、尿量減少等心功能不全表現,心功能Ⅱ級。該患兒存在的c.602T>C(p.I201T)變異已在多個LVNC 患者中檢出。研究發現I201T變異位于S1運動區,變異的產生可能會影響心肌的能量代謝[25]。Franaszczyk等[26]報道1 例19 歲DCM 合并LVNC 的男性患者存在MYH7基因c.602T>C 變異,心肌非致密層與致密層之比為3.2∶1,接受抗HF 治療后,24 歲時左心室功能仍低(LVEF 37%)。de Frutos 等[27]報道36% DCM 患者符合LVNC 影像學標準,男性確診年齡早于女性,隨訪5 年MACE 發生率為11.6%。本研究病例1 是女性患兒,末次隨訪年齡為30 個月,有家族史,患兒哥哥6 月齡時因CM 死亡。病例1 服用改善心功能的藥物后反應良好(LVEF 64%),但心肌重構未逆轉。提示性別可能對預后有一定影響[28],需定期隨訪,預防遠期預后不良事件發生。
綜上,兒童CM 基因型和表型間關聯并不明確。MYH7基因變異可導致不同的臨床表型,了解患兒的遺傳背景具有深遠的臨床意義。本研究通過分析5 例MYH7基因變異相關CM 患兒基因型與表型的特點,有助于臨床早期治療、遺傳咨詢及疾病預后評估。未來對于MYH7基因相關兒童CM機制的深入研究,將有望揭示MYH7基因型及表型關系,并為CM尋找更有效干預及精準治療方法。
利益沖突聲明:所有作者聲明無利益沖突。
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
少兒美術·書法版(2021年11期)2021-10-20 06:23:28
少兒美術·書法版(2021年8期)2021-10-20 06:08:10
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
汽車工程學報(2017年2期)2017-07-05 08:13:02
雜文選刊(2016年7期)2016-08-02 08:39:56
小天使·一年級語數英綜合(2016年6期)2016-05-14 12:21:05
