PPy對Al摻雜MnO2電極材料電化學性能的影響
2023-10-16 06:56:52李佳萌曾嘯雄談國強
陜西科技大學學報 2023年5期
關鍵詞:復合材料
夏 傲, 李佳萌, 曾嘯雄, 陳 軍, 談國強
(陜西科技大學 材料科學與工程學院 陜西省無機材料綠色制備與功能化重點實驗室, 陜西 西安 710021)
0 引言
超級電容器是一種電化學能源,是基于電化學能量轉換原理的非常規能源設備,其具有比電容高、充放電速度快、循環壽命長、穩定性好和環境友好等優點,有廣闊的應用前景,備受研究人員關注[1-4].
超級電容器的性能受到很多因素的影響,例如電極材料的電化學性能、電解質的選擇以及電極的電位窗口,電極材料的選擇在其中至關重要[5-8].因此,大量的研究工作致力于開發具有適當結構設計的超級電容器電極先進材料,以促進有效的電子傳輸和離子擴散[9].二氧化錳(MnO2)具有低成本、高比電容和環境兼容性,被廣泛用于電極材料,但作為半導體材料,MnO2的低電導率和低離子傳輸效率等缺點限制了它的實用性[10-13].
為了解決上述問題,研究人員提出了幾種改性方法:(1)提高電極的導電性[14];(2)使用納米結構,最大限度地減少擴散途徑,同時獲得更高的比表面積[15];(3)開發多孔材料以加速離子擴散[16].其中,向MnO2中摻雜金屬離子Al是一種有效的改性方法.Hu等[17]采用水熱法制備了Al摻雜的α-MnO2電極,比電容達到213 F/g,在15 000次循環后還能保持91%的循環穩定性.Huang等[18]通過化學沉淀方法合成了Al摻雜的MnO2,在1 A/g的電流密度下比電容為264.6 F/g,高溫比電容可達325.4 F/g,在室溫和50 ℃下都表現出良好的循環穩定性.此外,夏傲等[19]采用水熱法制備Al3+離子摻雜δ-MnO2,在1 A/g的電流密度下Al3+摻雜量為45%的電極(A0.45M)比電容為207.61 F/g,循環10 000次后比電容保持率為81.33%.單獨的離子摻雜雖然可以提高電極材料的電導率,但結構不夠穩定,MnO2易發生體積效應的問題還未得到解決.
聚吡咯(PPy)由于其良好的本征電導性和經濟性,在超級電容器中顯示出很好的應用前景,由于其快速的氧化還原反應、生物相容性以及較高的機械強度,在提高電化學性能方面備受關注[20,21].所以,通過復合材料設計將MnO2與PPy進行結合不僅可以提高超級電容器的電導率,還可以減小電極材料的體積效應.Wang等[22]通過模板法制備了MnO2@PPy復合材料,在1 A/g的電流密度下的比電容為295 F/g.組成的非對稱電容器在1 A/g的電流密度和0~2.2 V的電壓窗口下具有63 F/g的比電容,42 Wh/kg的能量密度和1 100 W/kg的功率密度.Zhou等[23]通過原位聚合法制備了核殼結構的PPy@MnO2復合材料,低電流密度下的比電容為614.7 F/g.與碳組成的非對稱電容器具有0~1.6 V的電壓窗口,34 Wh/kg的能量密度和317.6 W/kg的功率密度.然而,對于δ-MnO2先進行離子摻雜再與PPy復合的報道很少.
因此,本課題通過水熱法和低溫原位聚合法,制備Al3+摻雜MnO2與PPy的復合材料,研究復合量的變化對MnO2電極電化學性能的影響規律,得到了最佳的復合量,并深入分析PPy復合對δ-MnO2電化學性能提高的機理.
1 實驗部分
1.1 主要原料
高錳酸鉀(KMnO4),國藥集團化學試劑;鹽酸(HCl),國藥集團化學試劑;一水合硫酸錳(MnSO4·H2O),國藥集團化學試劑;六水合氯化鋁(AlCl3·6H2O),阿拉丁化學試劑;乙醇(C2H5OH),國藥集團化學試劑.吡咯(C4H5N),國藥集團化學試劑.泡沫鎳(Ni),云縱城科技有限公司.
1.2 Al摻雜MnO2的制備
用水熱法制備Al摻雜的MnO2.稱取0.19 g KMnO4、0.034 g MnSO4·H2O和0.022 g AlCl3·6H2O,溶于40 mL去離子水中.攪拌均勻后將溶液移入聚四氟乙烯內襯的高壓反應釜,在150 ℃下保溫16 h.反應結束后收集并洗滌產物,在70 ℃下干燥12 h,得到產物,命名為A0.45M.在本課題組之前的研究中,已對Al的摻雜量進行ICP-AES測試,得到A0.45M中實際摻雜量(摩爾分數)為0.56%[19].
1.3 Al摻雜MnO2復合PPy的制備
稱取一定量的A0.45M(0.270、0.231、0.193、0.154 g),量取20 mL濃度為0.1 mol/L的鹽酸,混合超聲30 min后加入0.04 mL吡咯單體(Py),攪拌至溶液顏色變黑,移入冰水浴中在0 ℃下反應4 h.將反映后的物質洗滌干凈,再置入70 ℃干燥箱干燥12 h,得到A0.45M/PPy復合材料.根據Py的用量將樣品分別記為A0.45M7P1、A0.45M6P1、A0.45M5P1、A0.45M4P1(A0.45M與Py的質量比分別為7∶1、6∶1、5∶1、4∶1).
1.4 樣品的形貌和結構表征
采用X射線衍射儀(XRD,D/max2200PC,日本理學株式會社)對樣品進行物相分析,Cu靶Kα為輻射源,波長λ=0.154 06 nm,管電壓40 KV,管電流100 mA,掃描角度10°~80°,掃速20°·min-1.利用場發射掃描電子顯微鏡(SU8100,日本HITACHI公司)與透射電子顯微鏡(TEM,FEI Tecnai G2 F20 S-TWIN,美國)對樣品進行形貌和微觀結構表征.通過傅里葉紅外光譜測試(FT-IR,VERTEX-70,德國)確定樣品的分子結構以及化學組成,測試波長為400~4 000 cm-1.通過熱重分析測試(TG,TGA Q500,美國)獲取材料組分的含量以及熱穩定性,升溫速率為10 ℃/min,升溫范圍為40 ℃~650 ℃.
1.5 電極的制備及電化學性能分析
稱取35 mg的活性物質,10 mg的導電碳粉,5 mg的PVDF(聚偏氟乙烯),依次放入瑪瑙研缽中充分混合研磨半小時后,加入適量分散劑NMP(N-甲基吡咯烷酮)充分研磨至具有一定粘度的漿料.將其均勻涂覆在裁剪成1×2 cm大小的泡沫鎳上,放入真空干燥箱中,70 ℃下干燥24 h.涂膜前與干燥后稱量記錄電極片,計算活性物質的量(活性物質負載量為0.85~1.00 mg·cm-2).
恒流充放電性能測試(GCD,0~1 V,1~10 A/g)、循環伏安測試(CV,0~1 V,5 mV/s)以及電化學阻抗測試(EIS,0.001~100 000 Hz)由電化學工作站(CHI 660E)完成.
2 結果與討論
2.1 XRD分析
圖1是A0.45M以及在其表面包裹了有機導電聚合物吡咯(PPy)的A0.45M/PPy復合材料樣品的XRD譜圖.從圖中可以看出,包裹PPy后,樣品的XRD譜圖相較于A0.45M沒有發生明顯變化,只是衍射峰強度有所下降.并且隨著PPy復合量的增加,衍射峰的強度逐漸降低.
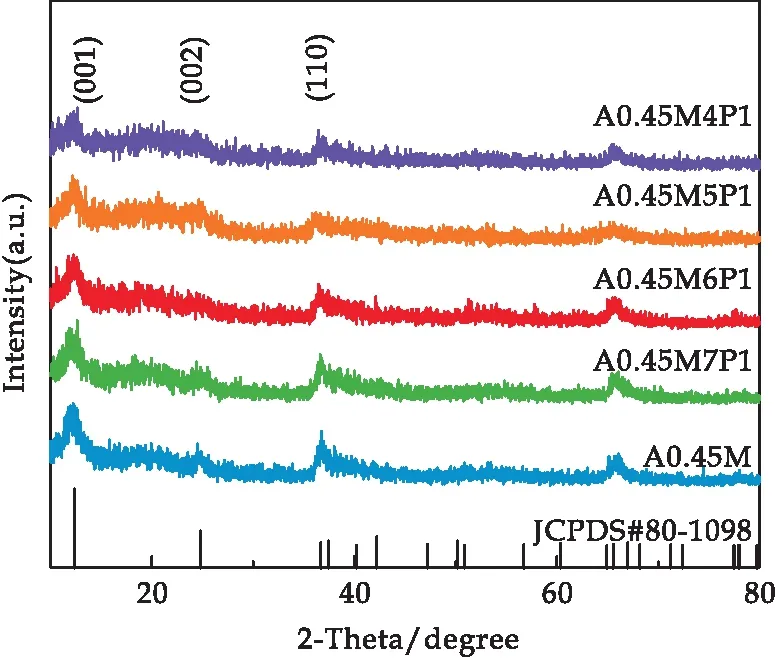
圖1 所有樣品的X射線衍射圖
2.2 SEM分析
圖2是A0.45M以及在其表面包裹了高分子導電聚合物吡咯(PPy)的A0.45M/PPy復合材料樣品的SEM照片.圖2(b)、(g)為A0.45M7P1的SEM照片,發現和A0.45M對應的圖2(a)、(f)SEM照片沒明顯變化,只是納米片的的厚度由12 nm增加到了16 nm,這可能是加入PPy較小,導致PPy對A0.45M包覆效果不明顯.
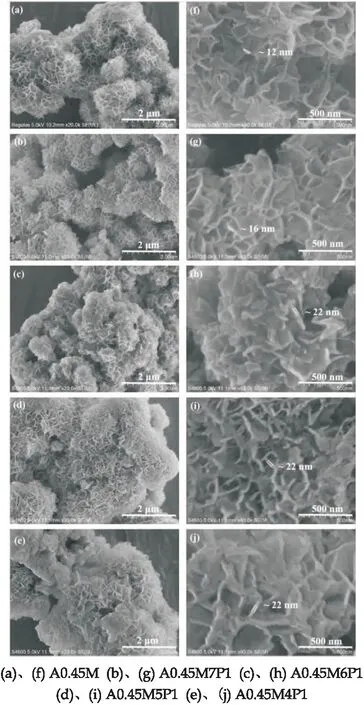
圖2 樣品的SEM圖
圖2(c)、(h)是A0.45M6P1的SEM照片,隨著PPy復合量的增大可以看出納米片明顯變厚,納米片的厚度達到22 nm并且花球孔洞有明顯的填充;圖2(d)、(i)為A0.45M5P1的SEM照片,隨著PPy復合量的進一步加大,可以看到納米片的厚度并沒有繼續增大依舊保持22 nm,但是A0.45M5P1花球組成的團簇發生了明顯的團聚,花球內部的空隙大小明顯變小;圖2(e)、(j)為 A0.45M4P1的SEM圖片,A0.45M4P1呈現的形貌與前幾種樣品都有所不同,產生了大量實心球和小部分花球,花球內部空隙也逐漸被填滿,導致了嚴重的團聚現象.團聚現象致使復合材料比表面積減小,活性位點隨之減少,導致性能下降.
2.3 TEM分析
TEM照片進一步揭示了樣品A0.45M6P1的微觀形貌.由圖3(a)可以看出,A0.45M6P1樣品還是成花球狀,花球直徑依舊為310 nm,但是花球之間的空隙相較于A0.45M變得更窄了.這主要是因為PPy的加入,與A0.45M完成了復合,PPy包裹住了表面的納米片,從而導致納米花球之間的空隙被填充.圖3(b)是高壓下TEM照片.通過EDS能譜表明,A0.45M6P1樣品中含有Mn、O、K、Al、C和N元素(圖3(c)~(h)).K離子在水熱過程中對δ-MnO2層狀結構的形成有著穩定結構的作用,對電化學反應無影響[24].C和N元素的均勻分布預示了PPy成功地與A0.45M完成了復合.
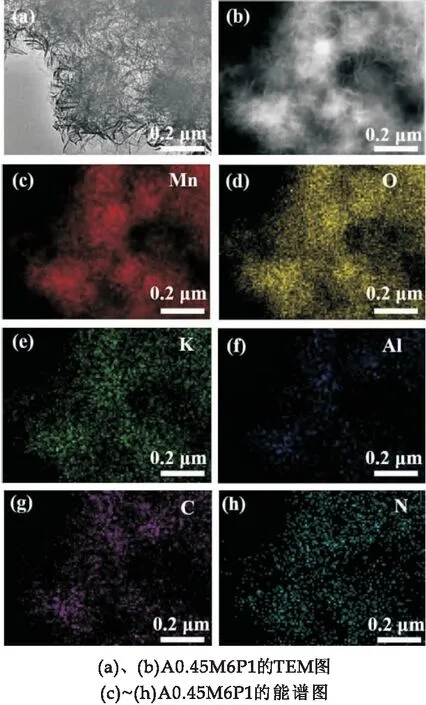
圖3 樣品的TEM圖
2.4 傅里葉紅外光譜分析(FT-IR)
采用FT-IR對A0.45M和A0.45M6P1復合材料進行化學鍵結構分析測試,其結果如圖4所示.其中,464 cm-1、510 cm-1、799 cm-1處出現的特征峰來源于MnO2中[MnO6]的Mn-O鍵伸縮振動[25,26].在1 010 cm-1、1 060 cm-1、1 093 cm-1和1 170 cm-1處出現的特征峰分別對應于=C-H鍵面外伸縮振動、N-H鍵面內伸縮振動、C-N鍵伸縮振動和=C-H鍵面內伸縮振動[23,25].1 400 cm-1、1 630 cm-1、1 740 cm-1處對應的特征峰分別對應于C-C鍵的伸縮振動、C=C鍵面內伸縮振動以及羰基/羧基相關的C=O鍵伸縮振動,而1 740 cm-1處出現的特征峰表明PPy與被包裹物質之間存在明顯的界面相互作用[23,26].經過對比可得PPy成功與A0.45M完成了復合.
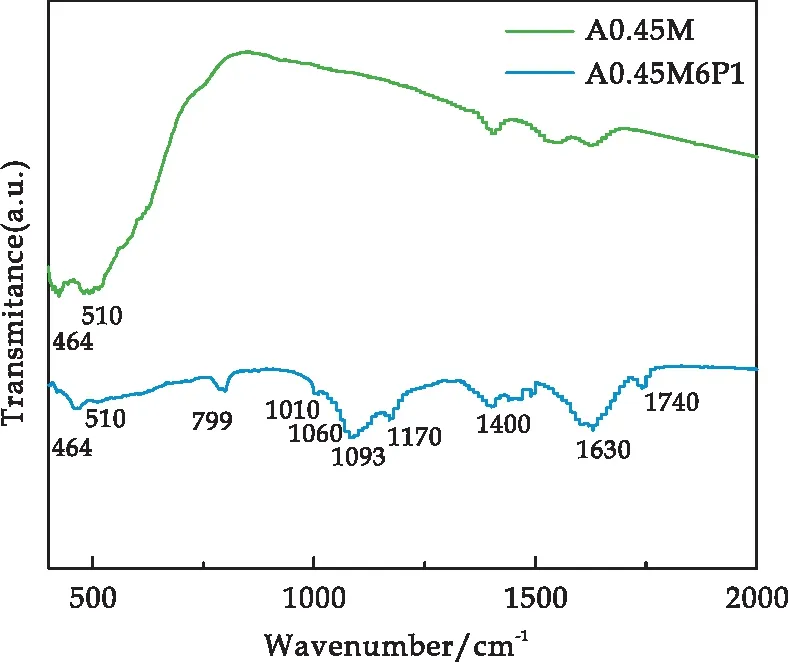
圖4 A0.45M和A0.45M6P1樣品的紅外光譜
2.5 熱重分析(TGA)
為確定A0.45M6P1復合材料中各組元的含量,對樣品進行TGA測試,其結果如圖5所示.在空氣氣氛下進行測試,升溫速率為10 ℃/min,升溫范圍為40 ℃~650 ℃.從圖中可以了解到,TGA曲線大致可以分為三個階段.第一個階段(40 ℃~205 ℃):A0.45M6P1樣品在205 ℃之前一共失重8.7%,該過程主要是為了除去樣品表面的吸附水和內部的結晶水;第二個階段(205 ℃~550 ℃):A0.45M6P1樣品又發生了9%的失重,這個階段主要是復合材料中的PPy的分子鏈受高溫而產生了熱分解;第三個階段(550 ℃~600 ℃):該過程發生了很明顯的失重現象,失重大小為1.69%,這主要對應MnO2的晶格失氧,Mn4+高溫還原為Mn3+從而產生氧氣先生成Mn2O3最終生成Mn3O4.TGA分析表明,A0.45M6P1復合材料中PPy的含量為9 wt%.
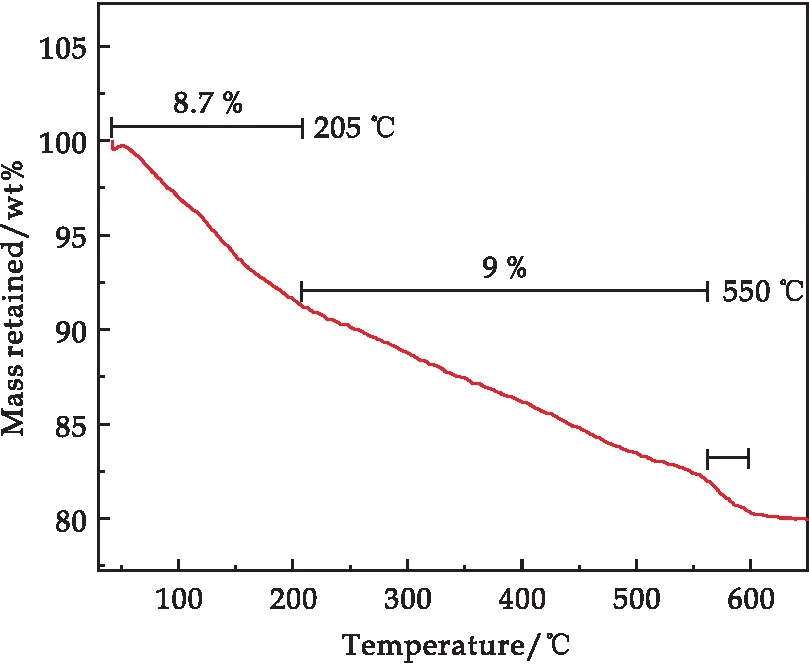
圖5 A0.45M6P1復合樣品的TGA譜圖
2.6 電化學性能分析
圖6(a)展示了不同樣品在5 mV/s掃描速率下的CV曲線.由圖可見,CV曲線呈矩形,表明電極具有理想的雙電層電容特性和快速反應動力學特性.由A0.45M、A0.45M6P1在不同掃描速度下的CV曲線可以看出,即使在50 mV/s高掃描速率下,A0.45M、A0.45M6P1的CV曲線仍保持準矩形(圖6(b)、(c)),說明樣品具有良好的可逆性和超快的充放電能力.A0.45M6P1的CV曲線比A0.45M面積更大,這意味著PPy的復合提高了MnO2的反應動力學.
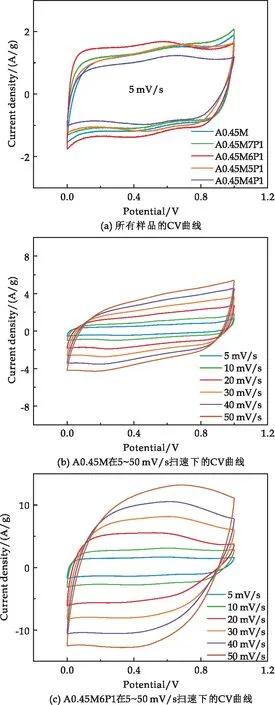
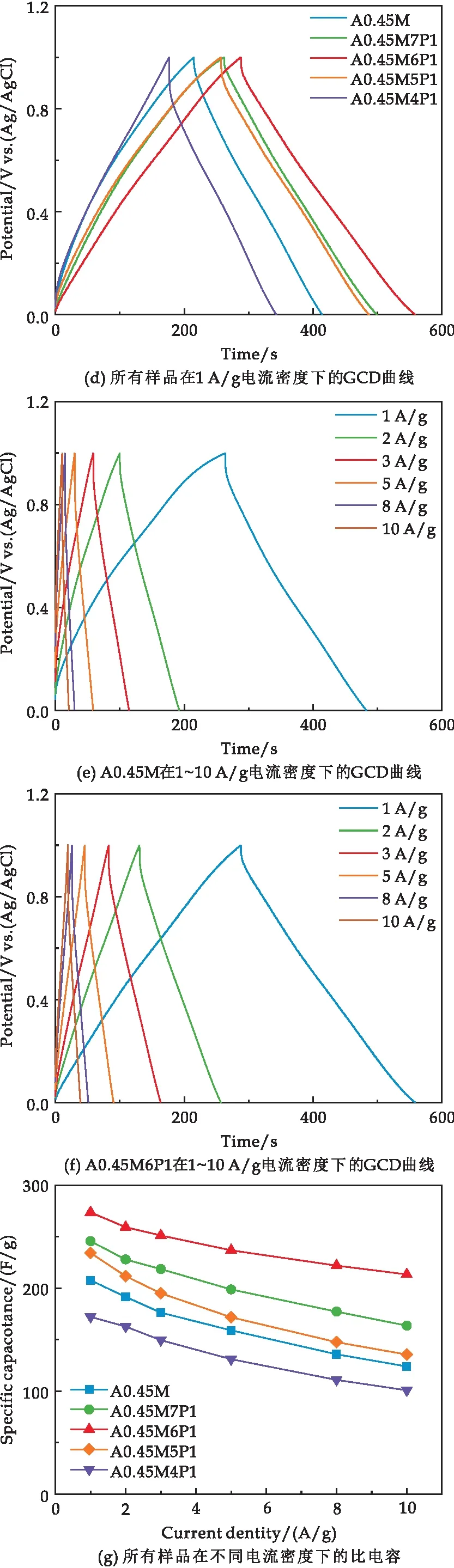
圖6(d)為5種樣品在1 A/g電流密度下的恒電流充放電曲線,所有曲線均呈等腰三角形.相比A0.45M,復合樣品的的放電時間延長、內部電壓(IR)降減小,其中A0.45M6P1的放電時間最長、IR降最小.IR降減小意味著樣品反應極化減小,反應可逆性增強.在1 A/g電流密度下,A0.45M、A0.45M7P1、A0.45M6P1、A0.45M5P1和A0.45M4P1的比電容分別為207.61 F/g、245.62 F/g、277.56 F/g、234.22 F/g和172.15 F/g.
圖6(e)、(f)分別為A0.45M和A0.45M6P1在1 A/g、2 A/g、3 A/g、5 A/g、8 A/g和10 A/g電流密度下的恒電流充放電曲線.隨著電流密度的增加,A0.45M和A0.45M6P1的充放電時間均縮短.然而,在相同電流密度下A0.45M6P1充放電時間約為A0.45M的1.3倍,說明A0.45M6P1的倍率性能顯著優于A0.45M.
圖6(g)對比了所有樣品在不同電流密度下的比電容.由圖可知,隨著電流密度(1、2、3、5、8、10 A/g)的增加,所有樣品的比電容呈下降趨勢,具體數值見表1所示.可知,相同電流密度下A0.45M6P1始終具有最高的比電容.電流密度從1 A/g增加至10 A/g時,A0.45M、A0.45M7P1、A0.45M6P1、A0.45M5P1和A0.45M4P1的比電容保持率分別為59.70%、66.60%、77.89%、57.84%和58.51%,對比可知適量的PPy復合可以有效提高MnO2的倍率性能,但過量的PPy會導致倍率性能下降,這與SEM的結果一致.
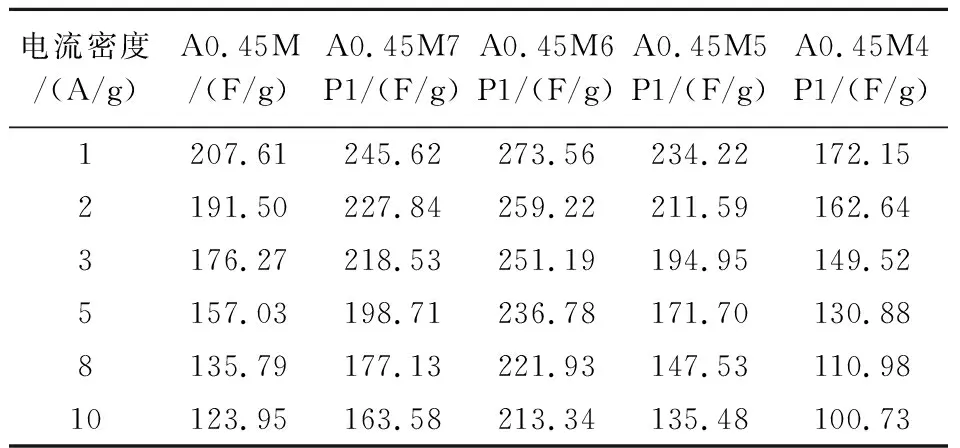
表1 所有樣品在不同電流密度下的比電容
圖6(h)為5種樣品在10 A/g電流密度下的循環性能測試結果.由圖可知,當PPy的復合量較低時,有助于A0.45M放電比電容的提升.隨著PPy的復合量不斷增大不僅不會提高A0.45M的放電比電容還會抑制A0.45M放電,降低A0.45M的放電比電容.其中A0.45M6P1的放電比電容最大.經過10 000次循環后,0.45M、A0.45M7P1、A0.45M6P1、A0.45M5P1和A0.45M4P1的放電比電容分別為100.81 F/g、153.62 F/g、196.78 F/g、109.57 F/g和65.07 F/g,比電容保持率分別為81.33%、94.33%、93.20%、82.80%和66.40%.PPy復合量較低時,對A0.45M的循環穩定性有顯著的提升,說明了A0.45M6P1具有良好的循環穩定性.
A0.45M6P1具有良好的電化學性能主要歸功于PPy為充放電過程中的電子提供了快速傳輸的路徑,在MnO2的納米片上復合上一層PPy有利于緩解MnO2在充放電過程中離子嵌入/脫出帶來的體積效應.
圖7(a)為所有樣品的EIS譜圖.在低頻段,A0.45M7P1、A0.45M6P1比A0.45M電極的斜率更陡峭而A0.45M5P1和A0.45M4P1比A0.45M的斜率傾斜度小,表明小復合量的PPy復合電極具有更理想的電化學雙電層電容行為,在電極表面具有更高的離子擴散速率和更低的擴散電阻(Zw),而大復合量的PPy會抑制電極的電化學性能.高頻段的EIS曲線都呈半圓弧型,數據擬合得到樣品A0.45M、A0.45M7P1、A0.45M6P1、A0.45M5P1和A0.45M4P1的Rct分別為2.28 Ω、1.94 Ω、0.69 Ω、2.73 Ω和5.93 Ω,可見低復合量的PPy可以降低A0.45M的Rct值,提高A0.45M的導電性.但是高復合量的PPy會增大A0.45M的Rct值,降低A0.45M的導電性.
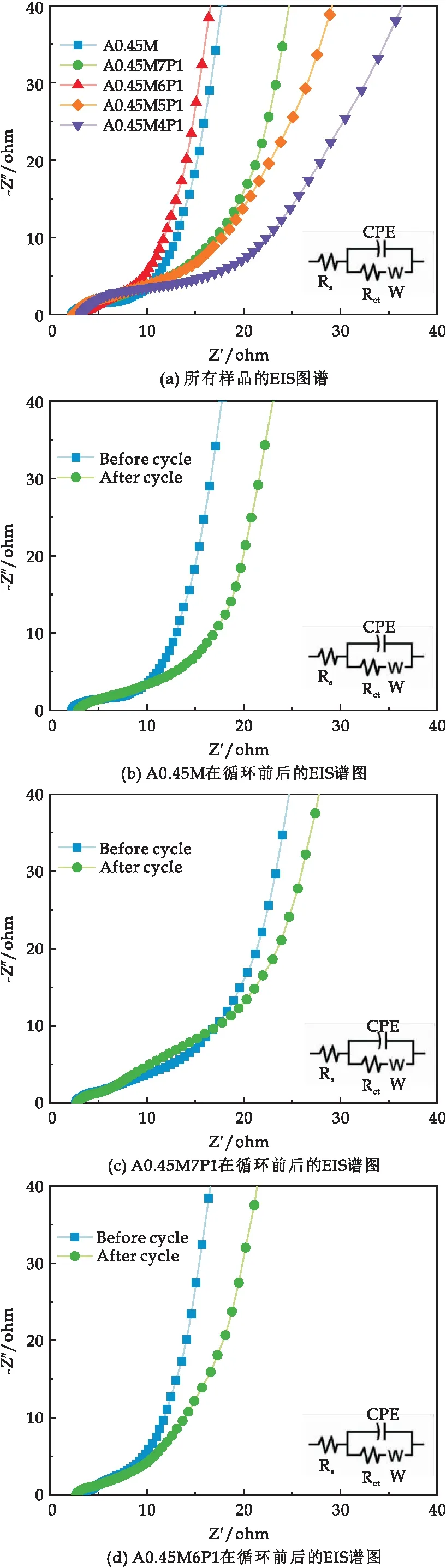
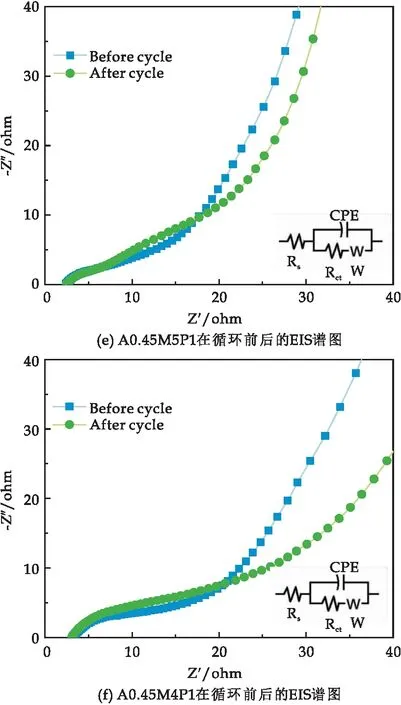
圖7 樣品的EIS譜圖
圖7(b)~(f)為5種樣品循環5 000次前后的EIS圖,分析得出所有樣品循環前后的Rct值如表2所示.結果表明,低復合量PPy的A0.45M的Rct值循環前后都明顯低于A0.45M,而高復合量PPy的A0.45M的 Rct值循環前后都高于A0.45M,這是因為在復合材料中,δ-MnO2作為支撐材料,為PPy的異質成核和均勻生長提供了豐富的活性位點,當PPy的復合量足夠大時,會發生額外的自聚合反應,形成蠕蟲狀團聚體,反而會降低導電性[27].除此之外,A0.45M6P1循環前后低頻區重合度最大,這說明了A0.45M6P1具有良好的結構穩定性.
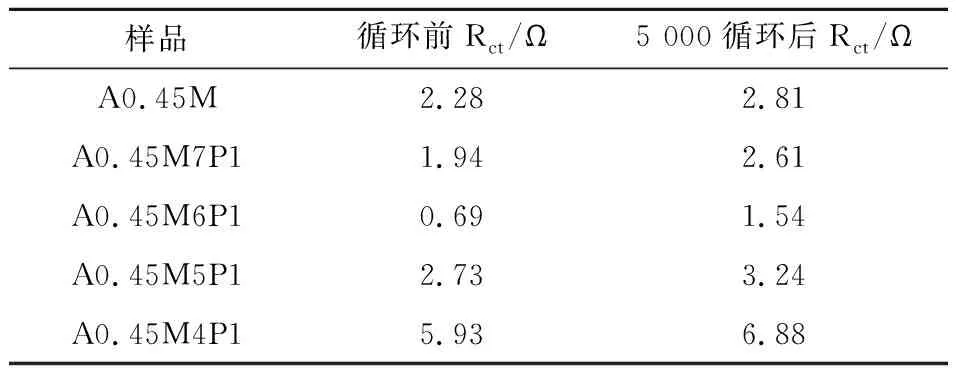
表2 樣品在循環前的EIS擬合數據以及在10 A/g的電流密度下循環5 000圈后的EIS擬合數據
在反應過程中PPy參與到電化學反應中,主要是由于PPy在Al摻雜的δ-MnO2(A0.45M)層狀結構上可控生長,改善了A0.45M的導電性,與此同時A0.45M提供較大的電容性能,為我們優化復合材料的電容性能提供了機會.為進一步探究A0.45M6P1復合材料的電荷存儲方式,根據A0.45M6P1在不同掃描速率(5~50 mV/s)的CV曲線(圖6(c))進行分析.在CV圖中,隨著掃描速率的增加,陽極和陰極的峰值傾向于向正負方向擴大.陽極和陰極的峰值電流如圖8(a)所示.峰值電流測量表明,電流響應與掃描速率線性相關[28].這不僅證明了電化學活性是由擴散控制的,而且在高掃描速率下也是可逆的[27].
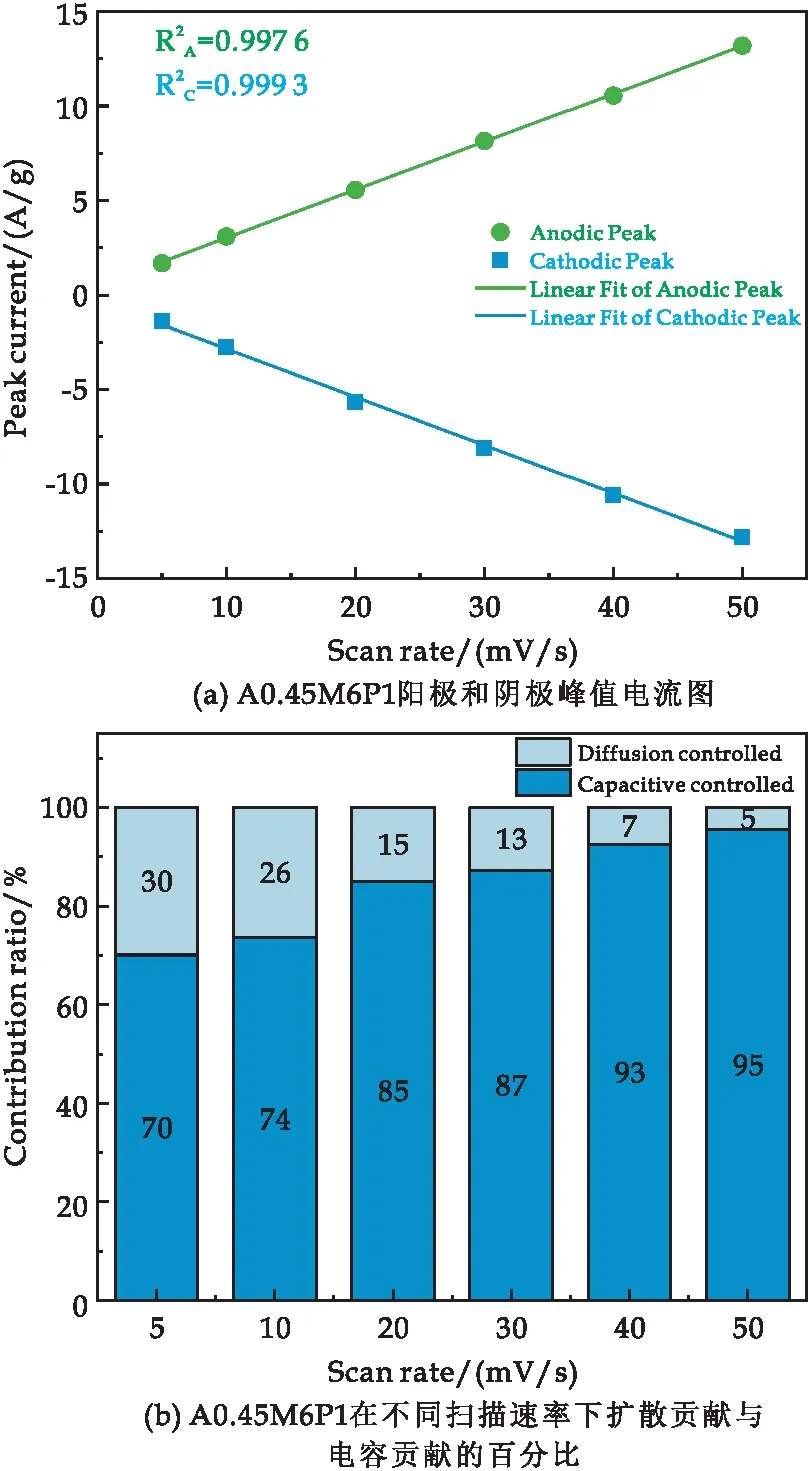
圖8 A0.45M6P1樣品的電化學性能圖
贗電容貢獻可按照公式(1)進行計算:
i(V)=k1v+k2v1/2
(1)
式(1)中:i為掃描速率為v時所得到的峰值電流大小,k1v代表了電容控制貢獻的電流、k2v1/2代表了擴散控制貢獻的電流.利用式(1)計算不同掃描速率下電容和擴散控制的貢獻率如圖8(b)所示.計算表明,當掃描速率較低時(5 mV/s),擴散控制貢獻達30%,隨著掃描速率的增加,電容控制貢獻逐漸增大,說明電容控制占主導地位.
3 結論
本文以Al摻雜MnO2(A0.45M)電極材料為基礎,通過低溫原位聚合法制備了PPy復合A0.45M的電極材料,研究了不同復合量的PPy對A0.45M電極材料的組成、結構、性能的影響,并進一步了解PPy對A0.45M電化學性能作用的機理.
結果表明,相較于A0.45M樣品,A0.45M6P1樣品具有更好的電化學性能.PPy可以緩解MnO2在充放電過程中的體積效應,也可以提高MnO2的導電性.在1 A/g、2 A/g、3 A/g、5 A/g、8 A/g、10 A/g的電流密度下時A0.45M6P1比電容分別為273.56 F/g、259.22 F/g、251.19 F/g、236.78 F/g、221.93 F/g和213.34 F/g,電流密度從1 A/g增加至10 A/g時比電容保持率為77.89%,表明PPy可以進一步地提高A0.45M的電化學性能.
猜你喜歡
建材發展導向(2022年2期)2022-03-08 01:44:04
建材發展導向(2021年14期)2021-08-23 00:56:16
中國材料進展(2019年10期)2019-12-07 05:32:14
纖維復合材料(2018年3期)2018-04-25 07:22:58
電子測試(2017年11期)2017-12-15 08:57:13
山東工業技術(2016年15期)2016-12-01 05:31:34
中國塑料(2015年6期)2015-11-13 03:02:54
中國塑料(2015年11期)2015-10-14 01:14:14
中國塑料(2015年8期)2015-10-14 01:10:41
應用化工(2014年10期)2014-08-16 13:11:29
