甲醇氣氛下低階煤熱解氣中CO加氫制芳烴機理研究
2023-01-04 09:23:10王月倫安會會張雪純馬云欣詹貴貴劉豪杰曹景沛
燃料化學學報 2022年12期
王月倫 ,安會會 ,張雪純 ,馬云欣 ,詹貴貴 ,劉豪杰 ,曹景沛
(1.中國礦業大學 煤炭加工與高效潔凈利用教育部重點實驗室, 江蘇 徐州 221116;2.中國礦業大學 江蘇省碳資源精細化利用工程研究中心, 江蘇 徐州 221116;3.徐州市建設工程檢測中心有限公司, 江蘇 徐州 221116)
輕質芳烴是重要的化工原料,廣泛用于醫藥、染料、香精及高分子行業,尤以苯、甲苯、二甲苯(簡稱BTX)市場需求更高。傳統的芳烴生產源于石油化工工藝,中國煤多油少的資源格局使煤炭的高效清潔利用日益受到重視[1,2]。熱解是低階煤分質利用的重點方向[3],熱解提質是優化煤熱解技術的有效途徑[4]。
低階煤熱解過程釋放大量的含氧芳香化合物、脂肪烴、熱解氣體等揮發分。這些組分可進一步轉化為輕質芳烴,因而以低階煤熱解揮發分重整制備輕質芳烴受到研究者關注[5-10]。煤熱解揮發物二次反應核心是C-O、C-C等鍵活化產生的自由基碎片與H·、CH3·等小分子自由基結合形成不同結構輕質產物。要實現活性物種有效匹配,熱解氣氛、催化劑是調控熱解行為和產物組成的關鍵。目前,多采用催化熱解以提高輕質芳烴選擇性,催化劑有金屬催化劑[5,7,9]、碳基催化劑[10]和分子篩催化劑[6-8]。其中,金屬改性分子篩催化劑因其雙活性中心協同使其成為重要的芳構化催化劑[11]。在催化過程中引入H2產生活性氫可穩定自由基碎片,提高輕質芳烴收率[12-14],但H2參與反應需高溫高壓,條件苛刻。外加供氫試劑通過氫轉移是實現C-C鍵、C-O鍵活化轉化的有效途徑。靳立軍等[15]采用甲烷芳構化與煤熱解耦合,由于甲烷能產生大量 H·、CH3·、CH2·和 CH·自由基,與煤熱解碎片耦合加氫顯著提高了低碳芳烴產率。
前期Wang等[16]研究了甲醇氣氛下低階煤熱解耦合揮發分原位重整制芳烴,發現甲醇氣氛能顯著提高輕質芳烴產率,歸因于甲醇在熱場生成了H·、CH3·等自由基。甲醇作為供氫試劑可實現C-C、C-O等鍵活化加氫,反應條件溫和易控[5,6]。文獻報道較多的是熱解揮發分中酚類和醚類組分轉化成單環芳烴[17,18]。由于低階煤含氧官能團多,熱解過程產生大量的CO和CO2,當體系存在活性氫,在催化劑作用下同樣可實現CO或CO2加氫生成輕質芳烴。目前,關于甲醇氣氛下低階煤熱解氣中CO或CO2制備輕質芳烴的研究鮮有報道,而近年來,以合成氣制備輕質芳烴引起了研究者廣泛關注[19,20]。根據反應中間體不同,合成氣制芳烴分為兩種途徑:一種為甲醇合成催化劑與分子篩耦合經中間產物甲醇制芳烴(SMA)[21];另一種為費托合成(FT)催化劑與分子篩耦合經過中間產物烯烴制芳烴(SOA)[22]。由于甲醇合成和甲醇芳構化反應條件不匹配,相比之下,費托合成反應和烯烴芳構化反應條件接近,更為適合合成氣一步法轉化為芳烴。與傳統的合成氣制芳烴不同的是本體系為甲醇氣氛下低階煤熱解氣中CO再轉化。CO可與熱解氣中的H2轉化制芳烴,同時甲醇經分子篩生成二甲醚,由C-C鍵偶聯后生成低碳烯烴(),再經齊聚、環化和氫轉移等也可轉化為芳烴。其中,環化脫氫產生的活性氫易使烯烴加氫形成烷烴。如活性氫能及時轉移,可避免烯烴加氫。研究發現[23],CO氣氛可有效促進芳構化反應。H-ZSM-5分子篩上甲醇芳構化產生的H可遷移至金屬活性中心,而H與吸附在金屬活性中心的CO進一步反應,加速脫氫芳構化發生。因此,甲醇脫氫芳構化耦合CO加氫有望促進芳烴的生成。該過程可解決低階煤熱解組分提質過程輕質芳烴產率低的問題,可為低階煤熱解獲取高附加值化學品提供技術支撐。目前,關于該過程制備輕質芳烴反應機理的理論計算鮮有報道。本工作基于Fe基催化劑對芳構化中間體烯烴有較高的選擇性,將過渡金屬Fe與HZSM-5分子篩耦合實現甲醇氣氛下煤熱解氣中CO加氫制芳烴。采用密度泛函理論(DFT)探討了CO在Fe/HZSM-5催化劑上加氫經由SOA過程生成芳烴可能的機理。
1 計算方法
鐵基催化劑在FT反應過程中相態結構存在變化,目前,已檢測到的活性相主要有金屬鐵、鐵的氧化物以及碳化物。研究表明,CO加氫活性中心主要為碳化鐵[23]。CO在Fe/HZSM-5催化劑上加氫制芳烴的反應可分為如下三步:在鐵基催化劑上生成低碳烯烴C2H4,反應活性相主要為碳化鐵(Fe5C2);低碳烯烴在HZSM-5中碳鏈增長生成;在HZSM-5中芳構化生成芳烴。
1.1 ZSM-5分子篩模型建立
由于小團簇模型具有原子個數少,對計算機配置要求低,計算量相對少等優點,且研究體系中參與反應的分子尺寸較小,在分子篩孔道中能與活性中心充分接觸,因此,本研究選取了如圖1所示由46個四面體單元組成的團簇模型ZSM-5,它包含了直孔道和正弦孔道以及孔道交叉處。ZSM-5分子篩的初始結構單元由Materials Studio數據庫導入,通過截取得到46T團簇模型。截取過程中形成的懸斷鍵由氫原子飽和,Si-H鍵長設置為1.47 ?,方向和初始模型中原來的Si-O鍵方向一致。ZSM-5分子篩的一個晶胞結構中含有12種不同的T位,鋁原子由于周圍環境的不同其穩定性不一樣,T12被認為較為穩定。當Al原子取代T12位所在的Si原子時,形成帶一個負電荷的四面體(),通過引入一個氫質子平衡此電荷,由此形成B酸中心位于孔道交叉處,作為反應的活性中心位置。
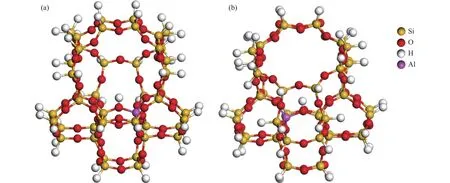
圖1 46T團簇模型示意圖Figure 1 Schematic diagram of 46T cluster model
1.2 Fe5C2 (510)模型建立
文獻報道采用Wulffconstruction方法可獲得Fe5C2微晶的平衡形貌及其暴露晶面的組成,表明Fe5C2(510)具有較低的表面能,且在暴露晶面中所占比例相對較高[24]。中間產物C2H4易于在Fe5C2(510)表面產生,因此,構建Fe5C2(510)模型。如圖2所示,該模型包括40個Fe原子和16個C原子,其中底部四層C原子和兩層Fe原子被固定,剩余的Fe原子和C原子位置弛豫。模型表面上真空層高度為 10 ?。計算時選用的模型大小為 P(2 × 1),k點值為(4 × 2 × 1),且考慮自旋極化效應。
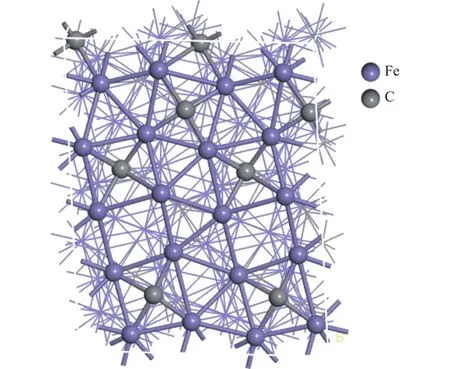
圖2 Fe5C2 (510)模型示意圖Figure 2 Schematic diagram of Fe5C2 (510) model
所有計算均在Materials Studio軟件包中CASTEP模塊下完成。計算過程中,設置平面波基組截斷動能為400 eV。第一布里淵區內的取樣采用了Monkhorst-Pack的網格,設置smearing的寬度為0.2 eV。電子的交換關聯能用廣義梯度近似(GGA)的PBE泛函來進行描述。過渡態是基于反應物和產物結構采用LST/QST方法搜索。
2 結果與討論
2.1 CO加氫生成C2H4過程
由CO加氫生成低碳烯烴C2H4包括CO解離、C1物種加氫、C1+ C1偶聯,涉及四步反應[25]。其反應熱、活化能、相對速率常數列于表1中。
CO解離:本研究計算采用CO直接解離機理。H和CO在Fe5C2(510)表面共吸附如圖3所示,H吸附在3F-3位,吸附能為-75 kJ/mol,CO吸附在4F-1位,吸附能為-140 kJ/mol,H和CO在Fe5C2(510)表面吸附均為最穩定構型。隨后CO和H開始擴散,CO通過TS1解離成C和O,圖4顯示的過渡態結構中,C-O鍵鍵長為1.96 ?。該步反應的活化能為124 kJ/mol,吸熱反應其反應熱為76 kJ/mol,其所需能壘最高,表明CO生成低碳烯烴決速步為C-O鍵的解離。

圖3 CO和H吸附構型圖Figure 3 CO and H adsorption configuration diagram
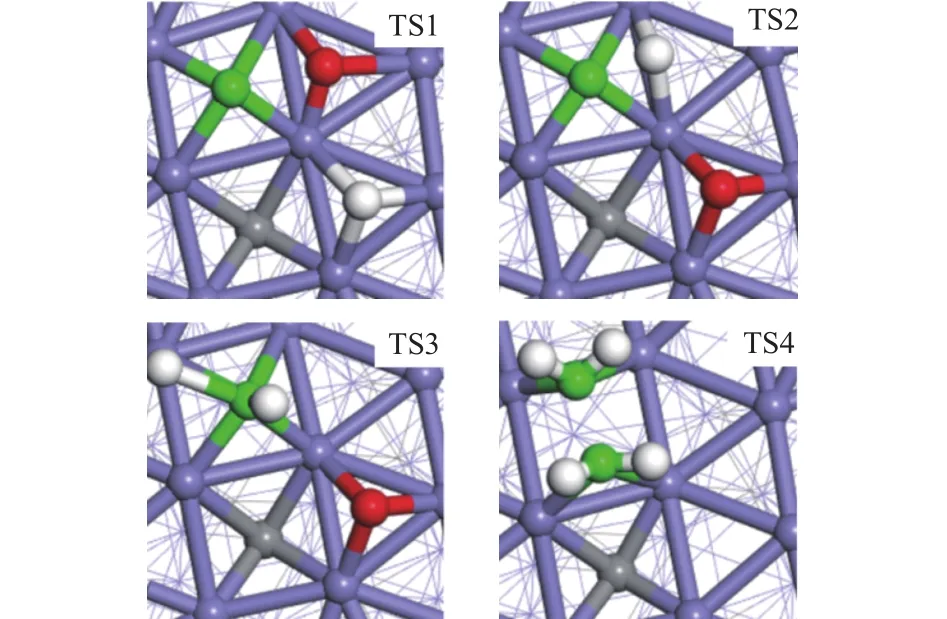
圖4 生成中間體C2H4過程中的過渡態Figure 4 Transition states in the formation of C2H4
C1物種加氫:C1逐步加氫生成CH和CH2,這兩步反應的活化能分別為12和82 kJ/mol,隨著活性氫數目增加,由于CH2相對于CH更不穩定,加氫需要的過渡態能壘更高,反應速率也隨之下降。在過渡態結構TS2和TS3中,即將形成的C-H鍵鍵長分別為1.43和1.92 ?。
C1+ C1偶聯:目前為止,文獻報道的鏈增長機制主要有三種:CO插入機理、碳化物機理和含氧中間體縮聚機理[26]。其中,碳化物機理已得到眾多理論與實驗驗證,該機理認為,CH2是C1+ C1偶聯鏈增長的單體。CH2偶聯過程中過渡態結構TS4中C-C鍵鍵長為1.787 ?,此步反應活化能為112 kJ/mol,是吸熱反應,反應熱為 10 kJ/mol。
能量變化如圖5所示,由圖5可知,生成中間體C2H4過程中的過渡態以TS4勢能最高,TS2勢能最低,表明C-C鍵偶聯實現碳增長最難,而活化后的C-O鍵再加氫形成CH中間體最容易。
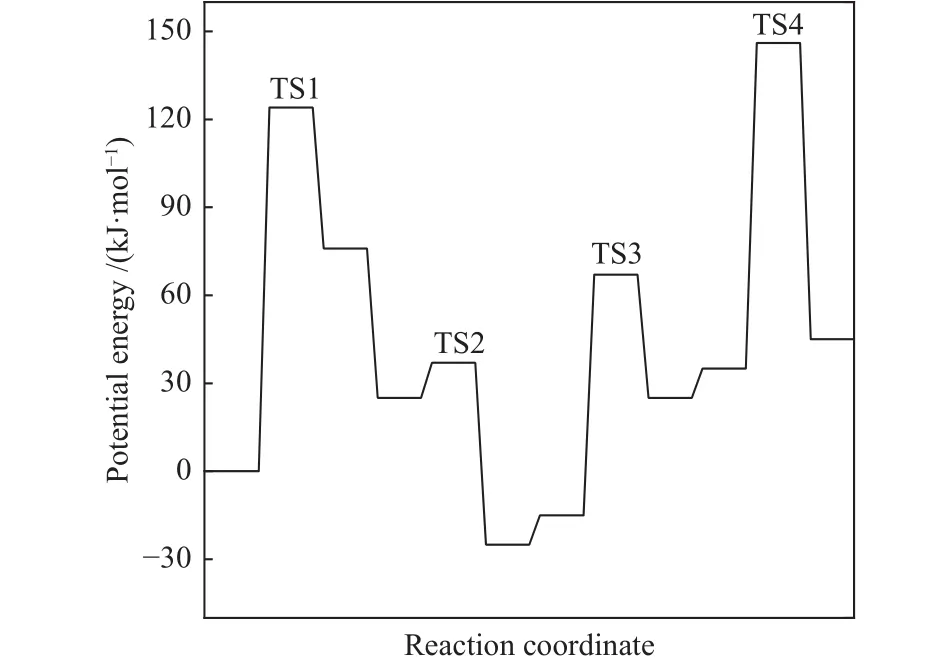
圖5 生成C2H4勢能圖Figure 5 Potential energies of the formation of C2H4
2.2 生成過程
甲醇氣氛下芳構化過程長鏈烯烴形成是由低碳烯烴甲基化得到,同時多甲基苯又可經過甲基化消去產生低碳烯烴,為長鏈烯烴的生成提供了烯烴的來源,因而仍然遵循雙循環機理[24]。圖6為低碳烯烴在HZSM-5催化劑中碳鏈增長生成的可能路徑。乙烯分子通過與甲醇不斷發生甲基化(M1、M2、M3和 M4)和去質子化(D1、D2和 D3)形成較高碳鏈的碳正離子,其經歷快速的分子內氫轉移和甲基轉移,最終生成己基碳正離子。
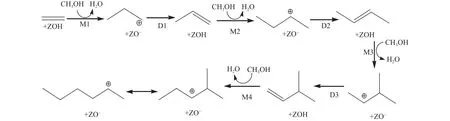
圖6 生成路徑示意圖Figure 6 Path of the formation of
表2 生成C計算結果Table 2 Calculation results of forming (823 K,101 kPa)
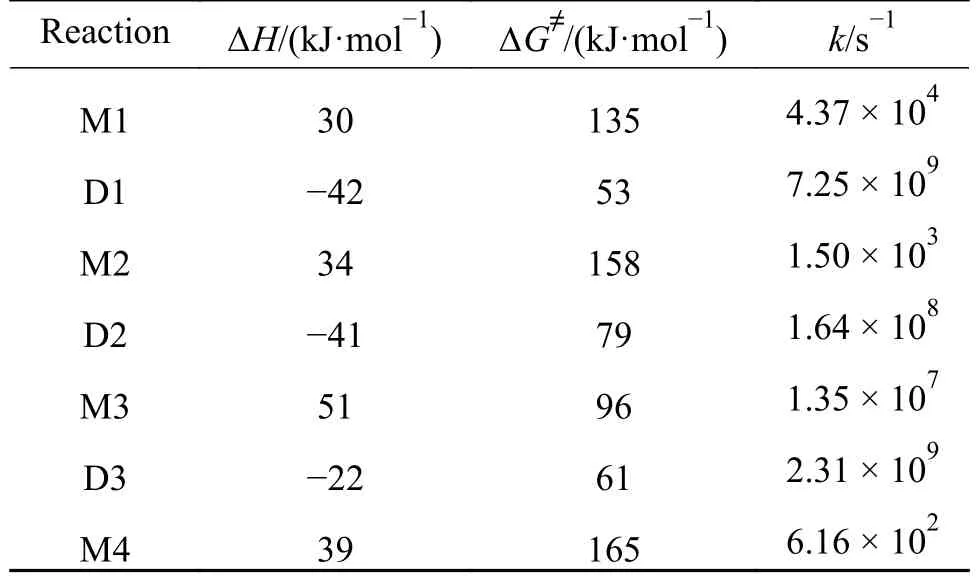
表2 生成C計算結果Table 2 Calculation results of forming (823 K,101 kPa)
Reaction ΔH/(kJ·mol-1) ΔG≠/(kJ·mol-1) k/s-1 M1 30 135 4.37 × 104 D1 -42 53 7.25 × 109 M2 34 158 1.50 × 103 D2 -41 79 1.64 × 108 M3 51 96 1.35 × 107 D3 -22 61 2.31 × 109 M4 39 165 6.16 × 102
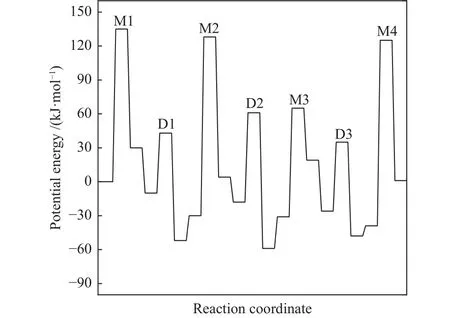
圖7 生成路徑勢能圖Figure 7 Potential energies of the formation of
甲基化:甲基化為乙烯和甲醇發生反應,脫去一分子水,生成碳正離子的過程[24]。四步甲基化的活化能分別為135、158、96與165 kJ/mol。甲基化過程速率常數值較低,表明反應速率較慢。
去質子化:碳正離子經歷去質子化形成新的烯烴產物。此過程為放熱反應,所需的活化能分別為53、79和61 kJ/mol,與甲基化過程相比,整體能壘降低,表明去質子化過程反應速率較快,容易發生。

圖8 芳構化路徑示意圖Figure 8 Process of aromatization for
2-己基碳正離子與分子篩發生迅速地去質子化過程(D1)形成1-己烯,甲醇脫水后形成的甲基吸附于分子篩氧原子上形成烷氧基,1-己烯與生成的烷氧基之間發生氫轉移(H1)得到烯烴碳正離子和甲烷,由于生成的烯烴碳正離子不穩定,發生快速的分子內氫轉移得到異構化之后的烯烴碳正離子,為接下來的環化提供了優化構型,從而發生環化反應(C1)生成環己碳正離子。環己烷碳正離子經歷同樣的去質子化(D2)過程后,與分子篩內甲氧基發生第二步氫轉移(H2)生成環己烯碳正離子。不穩定的環己烯碳正離子會繼續發生去質子化(D3)和第三步氫轉移(H3)生成環己二烯碳正離子,環己二烯碳正離子與分子篩發生最后一步去質子化(D4)生成了穩定產物苯。苯與甲醇不斷提供的甲基可進一步甲基化進而生成甲苯和二甲苯等多甲基苯。
表3 芳構化計算結果Table 3 Calculation results of aromatization(823 K,101 kPa)

表3 芳構化計算結果Table 3 Calculation results of aromatization(823 K,101 kPa)
Reaction ΔH/(kJ·mol-1) ΔG≠/(kJ·mol-1) k/s-1 D1 -18 57 4.13 × 109 H1 60 122 3.10 × 105 C1 -109 78 1.92 × 108 D2 -55 48 1.54 × 1010 H2 -29 107 2.77 × 106 D3 -11 31 1.85 × 1011 H3 -27 94 1.85 × 107 D4 -81 15 1.91 × 1012
整個去質子化過程均為放熱反應,所需的活化能在15-57 kJ/mol,其中,最小的速率常數為4.13 × 109s-1,表明在芳構化過程中,由不穩定的碳正離子去質子化形成己烯的反應相對容易,反應速率較快,而氫轉移過程所需能壘較高,反應速率相對較慢。
甲醇脫水分解為甲基的過程為:甲醇分子首先在分子篩B酸活性位吸附活化,導致甲醇的C-O鍵斷裂,然后形成甲基基團,并釋放一分子水。其脫水過程過渡態如圖10所示。此過程吸附的甲醇克服91 kJ/mol能壘脫水生成甲氧基,速率常數為2.87 × 107s-1,生成的水分子在下一步氫轉移過程前脫附。
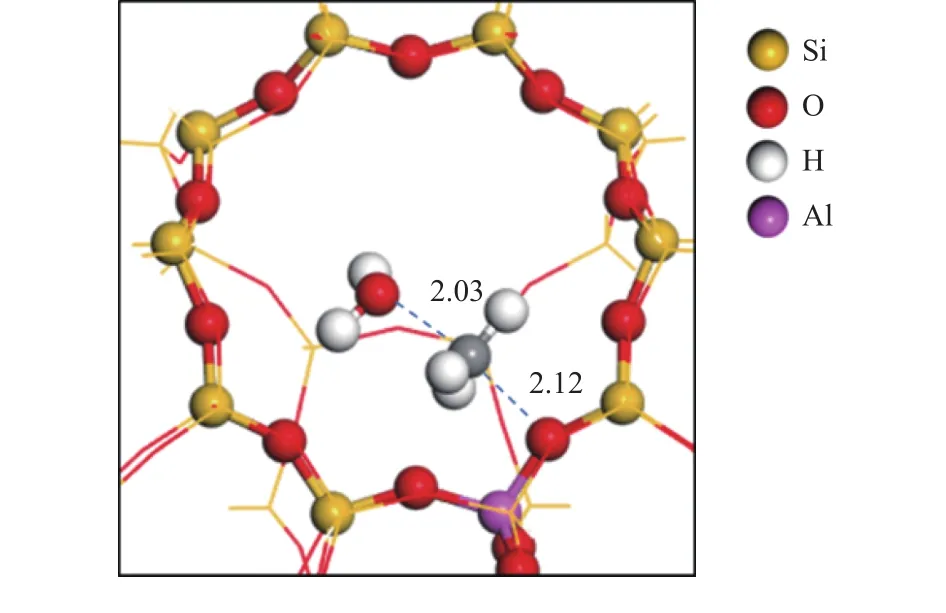
圖10 甲醇脫水過程過渡態Figure 10 Transition state of methanol dehydration process
氫轉移反應主要包括三步,第一步氫轉移(H1)發生于2-甲氧基與1-己烯之間,甲氧基從1-己烯分子中奪取一個氫原子,生成甲烷,1-己烯生成新的碳正離子。此步反應的活化能為122 kJ/mol,圖11所示的過渡態結構中,即將形成的CH4的C-H鍵鍵長為1.38 ?。第二步氫轉移(H2)生成環己烯碳正離子,該過程的活化能為107 kJ/mol。生成環己二烯碳正離子的第三步氫轉移(H3)的能壘為94 kJ/mol,速率常數為1.85 × 107s-1。在芳構化反應中,氫轉移過程所需要的活化能最高,反應速率最慢,可以看出氫轉移過程相對較難發生。在氫轉移過渡態結構中,即將形成的CH4的C-H鍵鍵長在1.38-1.61 ?,反應物分子即將斷裂的C-H鍵鍵長在1.35-1.38 ?,基本沒有變化。環化反應(C1)是指直鏈烯烴碳正離子環化生成環烷烴碳正離子,此反應的過渡態能壘為78 kJ/mol,為放熱過程其反應熱為109 kJ/mol。
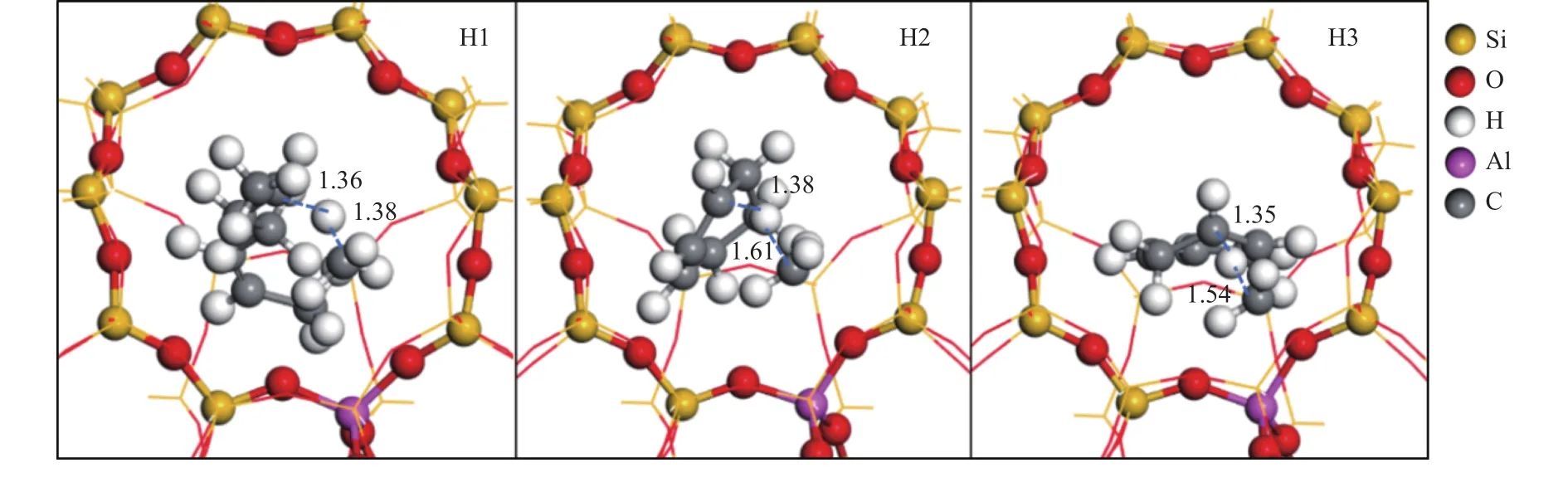
圖11 芳構化氫轉移過渡態Figure 11 Transition states of hydrogen transfer for aromatization
基于上述計算可知,相比傳統的合成氣制芳烴機理,H2在催化劑上吸附產生活性氫H,同時CO發生解離吸附進而與H生成CH2,CH2為鏈增長單體,通過不斷的C-C鍵偶聯實現鏈增長,隨后通過齊聚、環化和氫轉移形成芳烴。本研究為甲醇芳構化過程中脫氫,產生的活性氫與CO反應生成CH2,進而實現C-C鍵偶聯形成低碳烯烴,隨后與甲醇在分子篩上甲基化實現碳鏈增長,生成高碳離子,再環化脫氫芳構化生成芳烴。因而在此過程中甲醇不僅提供活性氫,還提供甲基。兩者耦合促進了輕質芳烴生成。
3 結 論
本研究采用DFT方法研究了甲醇氣氛下低階煤熱解氣CO于Fe/HZSM-5催化劑上加氫經烯烴中間體制芳烴機理。結果表明,甲醇氣氛下CO可于Fe5C2(510)表面加氫生成低碳烯烴,其CO解離為該過程的速控步。通過多次甲基化和去質子化過程C-C鍵偶聯實現鏈增長,其中,甲基化需較高的活化能。芳構化過程經過氫轉移、去質子化及環化(C)過程,其中氫轉移最難,反應速率常數最低。整個CO加氫制芳烴過程以甲基化所需能壘最高,成為該反應的決速步。總之,甲醇通過供氫或甲基為低階煤熱解氣中CO定向轉化制輕質芳烴提供了一條新途徑。
