新型含吡啶酮(吡唑)結構的A2a/A2b雙靶點腺苷受體拮抗劑的合成及生物活性研究
2022-10-11 14:03:16胡代強付信珍張淑敏
合成化學 2022年9期
李 志, 胡代強, 付信珍, 張淑敏*, 劉 明*
(1. 濱州醫學院 藥學院,山東 煙臺 264003; 2. 蘇州普瑞森生物科技有限公司,江蘇 蘇州 215004)
腫瘤的免疫治療在放療、化療和靶向藥物治療之后,成為治療腫瘤疾病的第三種革命性策略[1]。免疫治療以免疫檢查點抑制劑(Immune checkpoint inhibitors, ICI)[2]的臨床研究最為成熟,且應用最為廣泛[3]。ICI治療方法給癌癥患者帶來新的曙光,但是大量數據表明ICI治療只對部分患者有效,并且9%~29%的患者治療后會出現腫瘤“超進展”現象[4]。因此,如何激活、增強腫瘤的免疫治療應答率是腫瘤免疫療法走向廣泛應用的難題之一。
腺苷是腫瘤微環境中免疫應答的重要調節因子[5],其通過作用于腺苷受體(Adenosine Receptor,AR)發揮作用[6-7]。AR是一種G蛋白偶聯受體(Protein-Coupled Receptor, GPCR),其在藥理學中分為4類:A1、 A2a、 A2b和A3[8-9]。其中,A2aR尤為重要,是被廣泛研究的藥物靶點之一[10]。腺苷作為一種免疫抑制代謝物,可以與T細胞上表達的A2a受體結合,而這一結合會抑制T細胞的免疫反應,從而使腫瘤細胞產生“免疫逃逸”[11]。A2bR存在于樹突狀細胞中,在與腫瘤相關的巨噬細胞及骨髓來源的抑制性細胞中高表達[12]。而阻斷A2bR可以提高免疫細胞在腫瘤環境中的免疫應答,從而抑制腫瘤生長。A2受體拮抗劑可以與腺苷競爭性地結合A2a或A2b受體,進而保持相關免疫細胞的免疫活性,保留其殺傷腫瘤細胞的能力。目前,進入臨床的小分子腺苷受體拮抗劑主要包括:PBF-509[13-14]、 PBF-999、 CPI-444[15]、 CS3005和AB928[16]等。這些藥物無論是單用還是與PD(L)-1聯用,在腫瘤治療方面都取得了較好的臨床效果。值得注意的是,上述小分子腺苷受體拮抗劑大都只作用于A2aR單靶點,僅有AB928為A2a/A2b雙靶點藥物。
吡啶酮骨架作為一種兼有N-雜環和酰胺結構單元的化合物,在各種天然產物中都有發現[17-18]。其整合了多種小分子免疫檢查點抑制劑藥物的設計理念,在保留三唑聯嘧啶胺結構基礎上,引入了吡啶酮的結構,設計出了一類含有吡啶酮骨架的雙靶點A2腺苷受體拮抗劑。再根據生物電子等排原理,將這一類化合物衍生為含有吡唑結構的化合物,合成出了6個結構新穎的雙靶點A2腺苷受體拮抗劑(Scheme 1)。所得目標化合物均經1H NMR、13C NMR和HR-MS進行了結構確認,采用cAMP法[22]研究目標化合物對A2aR和A2bR的抑制活性。
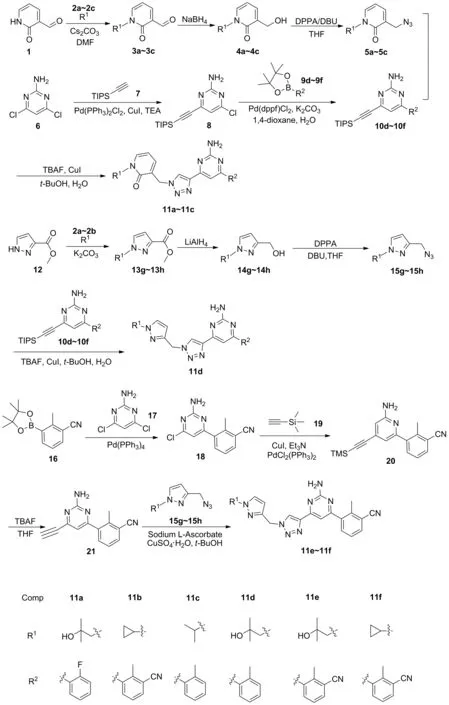
Scheme 1
1 實驗部分
1.1 儀器與試劑
X-4A型數字顯示顯微熔點儀;Bruker AVANCE NEO 600 MHZ型核磁共振儀(CDCl3或DMSO-d6為溶劑,TMS為內標);Thermo Q-Exactive型高分辨質譜儀;Waters QDa型液相色譜-質譜聯用儀(ESI電離模式);Waters e2695型高效液相色譜儀;Waters ACQUITY型超高效液相色譜儀。
中間體18、20和21按照文獻[20]方法合成;其余所用試劑均為分析純或化學純。
1.2 合成
(1)3a~3c的合成(以3a為例)
N2保護下,將化合物1(800 mg, 6.5 mmol)、甲基環氧丙烷(2a)(2.34 g, 32.5 mmol)、 Cs2CO3(6.35 g, 19.5 mmol)溶于N,N-二甲基甲酰胺(DMF)(8 mL)中,攪拌下于70 ℃反應12 h。反應結束后,依次用水和乙酸乙酯萃取3次。合并有機相,用飽和NaCl溶液洗滌、無水Na2SO4干燥、抽濾、減壓濃縮,殘余物經硅膠柱層析(洗脫劑:正己烷/乙酸乙酯=3/1,V/V)純化得化合物3a780 mg。
分別以環丙基硼酸(2b)、 2-碘代丙烷(2c)為原料,用類似的方法合成化合物3b、3c。
3a: 黃色固體,收率62%; MS(ESI)m/z: Calcd for C10H13NO3{[M+H]+}196.10, found 196.16。
3b: 淡黃色固體,收率61%; MS(ESI)m/z: Calcd for C9H9NO2{[M+H]+}164.07, found 164.12。
3c: 黃色固體,收率61%; MS(ESI)m/z: Calcd for C9H11NO2{[M+H]+}166.09, found 166.21。
(2)4a~4c的合成(以4a為例)
將化合物3a(780 mg, 4 mmol)溶于甲醇(10 mL)中,體系降溫至0 ℃,加入NaBH4(228 mg, 6mmol),于25 ℃反應1 h。反應結束后用水和乙酸乙酯萃取3次,合并有機相并減壓濃縮除去溶劑,得化合物4a788 mg。用類似的方法合成化合物4b和4c。
4a: 黃色油狀液體,收率100%; MS(ESI)m/z: Calcd for C10H15NO3{[M+H]+}198.11, found 198.21。
4b: 黃褐色油狀液體,收率97%; MS(ESI)m/z: Calcd for C9H11NO2{[M+H]+}166.09, found 166.24。
4c: 棕色油狀液體,收率100%; MS(ESI)m/z: Calcd for C9H13NO2{[M+H]+}168.10, found 166.31。
(3)5a~5c的合成(以5a為例)
N2保護下,將化合物4a(788 mg, 4 mmol)溶于15 mL無水四氫呋喃(THF)中,降溫至-5 ℃,依次滴加疊氮磷酸二苯酯(DPPA)(2.2 g, 8 mmol)和1,8-二氮雜二環十一碳-7-烯(DBU)(1.22 g, 8 mmol),在室溫下反應10 h。反應結束后用水和乙酸乙酯萃取,合并有機相,用飽和NaCl溶液洗滌,無水Na2SO4干燥,抽濾并減壓濃縮除去溶劑,殘余物經硅膠柱層析(洗脫劑:正己烷/乙酸乙酯=10/1,V/V)純化后得化合物5a400 mg。
用類似的方法合成化合物5b和5c。
5a: 黃色油狀液體,收率45%; MS(ESI)m/z: Calcd for C10H14N4O2{[M+H]+}223.12, found 223.18。
5b: 淡黃色油狀液體,收率51%; MS(ESI)m/z: Calcd for C9H10N4O{[M+H]+}191.09, found 191.21。
5c: 無色油狀液體,收率52%; MS(ESI)m/z: Calcd for C9H12N4O{[M+H]+}193.11, found 193.22。
(4)8的合成
N2保護下,依次將化合物6(16.4 g, 0.1 mol)、三異丙基硅基炔(7)(36.5 g, 0.2 mol)、Pd(PPh3)2Cl2(7.0 g, 0.01 mol)、 CuI(1.9 g, 0.01 mol)和三乙胺(30.3 g, 0.3 mol)加入THF溶液(300 mL)中,攪拌下于80 ℃反應過夜。反應結束后用乙酸乙酯和水萃取3次,合并有機相并減壓濃縮,粗產品經硅膠柱層析(洗脫劑 ∶二氯甲烷/甲醇=50/1,V/V)純化得20 g化合物8,淡黃色固體,收率64%; MS(ESI)m/z: Calcd for C15H24ClN3Si{[M+H]+}310.15, found 310.21。
(5)10d~10f的合成(以10d為例)
將化合物8(1.55 g, 5 mmol)、 2-氟苯基硼酸頻那醇酯(9d)(1.67 g, 7.5 mmol)、 Pd(dppf)Cl2(350 mg, 0.5 mmol)、 K2CO3(2.07 g, 15 mmol)溶于1,4-二氧六環(20 mL)和水(5 mL)的混合體系中。N2保護下,攪拌下于100 ℃反應4 h。反應結束后用乙酸乙酯和水萃取3次,合并有機相并減壓濃縮,粗產品經硅膠柱層析(洗脫劑:正己烷/乙酸乙酯=5/1,V/V)純化得1.2 g化合物10d。
分別以2-甲基-3-(4,4,5,5-四甲基-1,3,2-二氧雜硼雜環戊烷-2-基)苯甲腈(9e)、 2-甲基苯硼酸頻那醇酯(9f)為原料,用類似的方法合成化合物10e、10f。
10d: 淡黃色固體,收率65%; MS(ESI)m/z: Calcd for C21H28FN3Si{[M+H]+}370.21, found 370.14。
10e: 淡黃色固體,收率77%; MS(ESI)m/z: Calcd for C23H30N4Si{[M+H]+}391.23, found 391.17。
10f: 黃色固體,收率82%; MS(ESI)m/z: Calcd for C22H31N3Si{[M+H]+}366.24, found 366.31。
(6)11a~11c的合成(以11a為例)
將化合物5a(370 mg, 1 mmol)、化合物10d(333 mg, 1.5 mmol)、碘化亞銅(25 mg, 0.1 mmol)、四丁基氟化銨(TBAF)(523 mg, 2 mmol)溶于t-BuOH(4 mL)和水(4 mL)中,攪拌下于60 ℃反應過夜。反應結束后用乙酸乙酯和水萃取,合并有機相,減壓。粗產品經硅膠柱層析純化得30 mg化合物11a。
分別以5b、10e和5c、10f為原料,用類似的方法合成化合物11b、11c。
11a: 白色固體,收率51%, m.p.225~226 ℃;1H NMR(600 MHz, DMSO-d6)δ: 8.55(s, 1H), 8.02(td,J=7.8 Hz, 1.7 Hz, 1H), 7.67(dd,J=6.8 Hz, 2.1 Hz, 1H), 7.59(d,J=2.3 Hz, 1H), 7.56(m,J=7.8 Hz, 6.9 Hz, 5.1 Hz, 1.9 Hz, 1H), 7.49(dd,J=6.9 Hz, 2.0 Hz, 1H), 7.46~7.20(m, 2H), 6.84(s, 2H), 6.26(t,J=6.8 Hz, 1H), 5.48(s, 2H), 4.77(s, 1H), 3.94(s, 2H), 1.06(s, 6H);13C NMR(151 MHz, DMSO-d6)δ: 163.32, 160.69, 160.64, 160.57, 160.56, 159.03, 157.71, 144.94, 140.29, 138.83, 131.55, 131.49, 129.74, 129.73, 124.79, 124.71, 124.38, 124.20, 124.14, 124.12, 115.93, 115.78, 104.17, 104.11, 103.43, 69.24, 56.34, 49.17, 26.76; HR-MS(ESI)m/z: Calcd for C22H22N7O2F{[M+H]+}436.1892, found 436.1888。
11b: 白色固體,收率49%, m.p.161~162 ℃;1H NMR(600 MHz, DMSO-d6)δ: 8.60(s, 1H), 7.89(dd,J=7.7 Hz, 1.4 Hz, 1H), 7.75(dd,J=7.8 Hz, 1.3 Hz, 1H), 7.61(dd,J=7.0 Hz, 2.0 Hz, 1H), 7.55~7.49(m, 1H), 7.46(dd,J=6.9 Hz, 2.0 Hz, 1H), 7.25(s, 1H), 6.91(s, 2H), 6.24(t,J=6.9 Hz, 1H), 5.48(s, 2H), 3.38(tt,J=7.5 Hz, 4.2 Hz, 1H), 2.54(s, 3H), 1.03~0.95(m, 2H), 0.88~0.81(m, 2H);13C NMR(151 MHz, DMSO-d6)δ: 166.03, 163.01, 161.49, 157.62, 144.91, 139.40, 138.46, 138.29, 137.52, 133.00, 132.64, 126.34, 124.62, 124.14, 117.39, 112.58, 104.26, 104.04, 48.98, 31.37, 17.64, 5.76; HR-MS(ESI)m/z: Calcd for C23H20N8O{[M+H]+}425.1833, found 425.1827。
11c: 白色固體,收率54%, m.p.218~219 ℃;1H NMR(600 MHz, DMSO-d6)δ: 8.57(s, 1H), 7.81(dd,J=7.0 Hz, 2.0 Hz, 1H), 7.43(dd,J=7.5 Hz, 1.5 Hz, 1H), 7.41(dd,J=6.9 Hz, 1.9 Hz, 1H), 7.35(td,J=7.4 Hz, 1.5 Hz, 1H), 7.33~7.27(m, 2H), 7.23(s, 1H), 6.77(s, 2H), 6.33(t,J=6.9 Hz, 1H), 5.47(s, 2H), 5.09(hept,J=6.8 Hz, 1H), 2.39(s, 3H), 1.29(d,J=6.8 Hz, 6H);13C NMR(151 MHz, DMSO-d6)δ: 167.71, 163.01, 159.62, 157.11, 145.15, 138.05, 137.76, 134.67, 134.42, 130.10, 128.27, 128.21, 125.25, 124.39, 124.30, 104.73, 104.20, 49.13, 45.72, 20.77, 19.43; HR-MS(ESI)m/z: Calcd for C22H23N7O{[M+H]+}402.2037, found 402.2031。
(7)13g和13h的合成(以13g為例)
將化合物7(1.00 g, 7.93 mmol)加入100 ml三口圓底燒瓶中,用15mL DMF溶解,依次加入K2CO3(2.19 g, 15.86 mmol)和1-氯-2-甲基-2-丙醇(2a)(1.12 g, 10.31 mmol),于80 ℃反應17 h。用水和乙酸乙酯萃取并合并有機相,用飽和NaCl溶液萃取,無水Na2SO4干燥,抽濾并減壓濃縮除去溶劑,殘余物經硅膠柱層析(洗脫劑 ∶正己烷/乙酸乙酯=3/1,V/V)純化得0.617 g化合物13g。
以環丙基硼酸(2b)為原料,用類似的方法合成13h。
13g: 白色固體,收率39%;1H NMR(400 MHz, CDCl3)δ: 7.58(d,J=2.1 Hz, 1H), 6.97(d,J=2.1 Hz, 1H), 4.28(s, 2H), 1.51(s, 6H); MS(ESI)m/z: Calcd for C9H14N2O3{[M+H]+}199.11, found 199.38。
13h: 類白色固體,收率56%; MS(ESI)m/z: Calcd for C8H10N2O2{[M+H]+}167.08, found 167.28。
(8)14g和14h的合成(以14g為例)
N2保護下,將LiAlH4(115 mg, 3.03 mmol)溶解于10 ml無水THF。降溫至0 ℃,將化合物13g(200 mg, 1.01 mmol)的無水THF溶液轉移至上述體系,室溫攪拌3 h。反應結束后,將體系降溫至0 ℃,用2M氫氧化鈉溶液0.5 mL淬滅反應。用水和乙酸乙酯萃取3次。合并有機相,用飽和NaCl溶液洗滌,無水Na2SO4干燥、抽濾并減壓濃縮除去溶劑得150 mg化合物14g。
以13h為原料,用類似的方法合成14h。
14g: 無色油狀液體,MS(ESI)m/z: Calcd for C8H14N2O2{[M+H]+}171.11, found 171.28;直接進行下一步反應。
14h: 淡黃色液體,MS(ESI)m/z: Calcd for C7H10N2O{[M+H]+}139.09, found 139.12;直接進行下一步反應。
(8)15g和15h的合成
分別以14g、14h為原料,類似于化合物5a的合成步驟,得化合物15g、15h。
15g: 淡黃色液體,收率73.3%;MS(ESI)m/z: Calcd for C8H13N5O{[M+H]+}196.12, found 196.28。
15h: 淡黃色液體,收率66%;MS(ESI)m/z: Calcd for C7H9N5{[M+H]+}164.09, found 164.26。
(9)11d的合成
以化合物15g和10e為原料,類似于化合物11a的合成步驟,得化合物11d。
11d: 白色固體,收率10%, m.p.85~86 ℃;1H NMR(600 MHz, DMSO-d6)δ: 8.51(s, 1H), 7.81(d,J=0.8 Hz, 1H), 7.54(d,J=0.7 Hz, 1H), 7.43(dd,J=7.5 Hz, 1.5 Hz, 1H), 7.35(td,J=7.4 Hz, 1.5 Hz, 1H), 7.33~7.27(m, 2H), 7.22(s, 1H), 6.73(s, 2H), 5.56(s, 2H), 4.68(s, 1H), 3.99(s, 2H), 2.39(s, 3H), 1.04(s, 6H);13C NMR(151 MHz, DMSO-d6)δ: 167.69, 162.98, 157.10, 145.46, 138.02, 137.40, 134.68, 130.44, 130.10, 128.27, 128.22, 125.25, 123.15, 114.25, 104.27, 68.54, 61.42, 43.46, 26.55, 19.43; HR-MS(ESI)m/z: Calcd for C21H24N8O{[M+H]+}405.2146, found 405.2139。
(10)11e和11f的合成(以11e為例)
25 ℃下,將化合物20(100 mg,0.50 mmol)加入50 mL三口圓底燒瓶中。加入1 mL DMF、 5 mL叔丁醇和3 mL水,攪拌溶解。依次加入化合物15g(106 mg,0.55 mmol)、五合水硫酸銅(2.5 mg, 0.01 mmol)和L-抗壞血酸鈉(10 mg, 0.05 mmol),60 ℃攪拌過夜。轉移至單口瓶濃縮除去溶劑,硅膠柱層析(洗脫劑:二氯甲烷/甲醇=25/1,V/V)得150 mg化合物11e。
以化合物20和15h為原料,用類似方法合成化合物11f。
11e: 類白色固體,收率41%, m.p.173~174 ℃; 1H NMR(600 MHz, DMSO-d6)δ: 8.51(s, 1H), 7.89(dd,J=7.7 Hz, 1.4 Hz, 1H), 7.75(dd,J=7.7 Hz, 1.4 Hz, 1H), 7.65(d,J=2.3 Hz, 1H), 7.53~7.49(m, 1H), 7.25(s, 1H), 6.87(s, 2H), 6.29(d,J=2.2 Hz, 1H), 5.65(s, 2H), 4.67(s, 1H), 3.99(s, 2H), 2.54(s, 3H), 1.04(s, 6H);13C NMR(151 MHz, DMSO-d6)δ: 167.10, 164.06, 158.64, 146.31, 145.67, 140.45, 139.55, 134.09, 133.72, 133.06, 127.42, 124.77, 118.47, 113.66, 105.42, 104.97, 69.61, 62.42, 47.77, 27.64, 18.71; HR-MS(ESI)m/z: Calcd for C22H23N9O{[M+H]+}430.2098, found 430.2097。
11f: 淡黃色固體,收率53%, m.p.166~167 ℃; 1H NMR(600 MHz, DMSO-d6)δ: 8.53(s, 1H), 7.89(dd,J=7.7 Hz, 1.3 Hz, 1H), 7.78~7.73(m, 2H), 7.51(t,J=7.7 Hz, 1H), 7.25(s, 1H), 6.87(s, 2H), 6.26(d,J=2.3 Hz, 1H), 5.62(s, 2H), 3.70(tt,J=7.4 Hz, 3.9 Hz, 1H), 2.54(s, 3H), 1.02~0.93(m, 4H);13C NMR(151 MHz, DMSO-d6)δ: 167.10, 164.07, 158.62, 146.34, 146.23, 140.45, 139.55, 134.09, 133.72, 131.75, 127.42, 124.91, 118.47, 113.66, 105.44, 105.14, 47.77, 33.14, 18.71, 6.71; HR-MS(ESI)m/z: Calcd for C21H19N9{[M+H]+}398.1836, found 398.1835。
1.3 活性測試
采用cAMP法分別測定合成的化合物對A2a和A2b受體的抑制活性。實驗分別使用CHO-K1/ADORA2a/Gα15穩定轉染細胞系和CHO-K1/ADORA2b/Gα15穩定轉染細胞系[20]。
1.4 分子對接
利用Discovery Studio 2017 R2軟件,選取人源A2a腺苷受體A2aR-StaR2-bRIL(PDB ID:5IU4)[21]作為受體模型,A2b腺苷受體的蛋白模型是基于(PDB ID: 6PS7)[22]的同源模型,序列同一性61.92%。利用軟件中的CDOCKER模塊進行分子對接研究。
2 結果與討論
2.1 合成
實驗過程中,分別對化合物18、13g和11a~11d的實驗條件作了進一步優化,結果如表1~3所示。化合物18的合成中,K2CO3的當量由3倍當量增加至5倍當量,堿性增強,有利于催化劑四(三苯基膦)鈀(Pd(PPh3)4)的催化活性提高,從而使反應收率得到了提高。化合物13g的合成中,隨著反應溫度的提高,反應體系中分子之間的碰撞愈加劇烈,有利于反應收率的提高,但當溫度升高至95 ℃,化合物12產生了異構現象,并發生了自身的分子內成環反應。化合物11a~11d的合成中,一價銅離子是催化反應發生所必需的。L-抗壞血酸鈉作為配體,先將五合水硫酸銅中的二價銅離子還原為一價銅離子,再催化反應的發生。故直接使用碘化亞銅有利于反應收率的提高。

表1 化合物18 優化條件及實驗結果
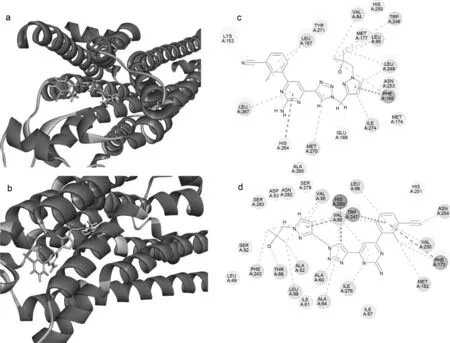
圖1 (a)化合物11e 與A2aR的結合模式;(c)分子對接結果二維作用模式;(b)化合物11e 與A2bR的結合模式;(d)分子對接結果二維作用模式

表2 化合物13g 優化條件及實驗結果

表3 化合物11a~11d 優化條件及實驗結果
2.2 活性測試
采用cAMP法在3種不同濃度下測定了合成的6個化合物對A2a受體的抑制活性并計算抑制率,以AB928做陽性對照,結果見表4。 6個化合物對A2aR受體均表現出不同程度的抑制活性。在1000 nM濃度時,11b和11e的抑制率最強,分別為96.31%、 96.08%。在100 nM濃度時,11b和11f對A2aR受體的抑制率高于陽性藥AB928。在10 nM濃度時,11b、11e和11f和均顯示出了較好的抑制活性,抑制率均高于陽性藥AB928。同時,通過對比抑制率,如11c和11d,初步推斷目標化合物中含有吡啶酮結構有利于活性的提高。選取了11b、11e和11f三個目標化合物,分別測定了抑制A2aR和A2bR的IC50值,結果如表5所示。其中,化合物11e抑制A2aR的IC50值(8.188 nM)與陽性藥AB928相當,抑制A2bR的IC50值(15.22 nM)明顯小于陽性藥AB928。

表4 化合物對A2a受體的抑制活性

表5 化合物抑制A2aR和A2bR受體的IC50值
2.3 分子對接
為了明確目標化合物與A2a(PDB ID: 5IU4)、 A2b之間的相互作用,使用Discovery Studio 2017 R2軟件對化合物11e進行對接研究。對接結果顯示,化合物11e與A2a(40.2565)、A2b(36.2381)對接的CDOCKER INTERACTION ENERGY均高于陽性藥物AB928(35.3138),說明化合物11e與A2a和A2b靶點具有較好的親和作用。化合物11e結構中的氰基、亞甲基除了通過范德華力分別與A2aR的LYS153、GLU169相互作用外,還與LEU249、 VAL84、 ILE274等殘基相互作用。另外,化合物11e結構中的氰基、羥基還可以分別與A2bR的ASN254、 THR89通過氫鍵相互作用提高與靶標的結合能力。
設計并合成了一系列具有吡啶酮或吡唑結構的A2腺苷受體拮抗劑,采用cAMP法研究化合物對A2aR和A2bR受體的抑制活性,并利用分子對接模擬化合物11e對A2a和A2b靶點的結合情況。結果表明:化合物11e對A2aR和A2bR的抑制活性最強,其中對A2bR的抑制性優于陽性對照藥AB928。化合物11e是一個具有廣泛前景的雙靶點(A2a和A2b)腺苷受體拮抗劑,也為小分子免疫檢查點抑制劑的研究與開發提供了有價值的借鑒意義。
