馬來酸氟伏沙明-D3的合成
2022-10-11 14:03:20仇中選黃龍江
合成化學 2022年9期
仇中選, 王 東, 黃龍江
(1. 青島職業技術學院 生物與化工學院,山東 青島 266555; 2. 青島科技大學 化工學院,山東 青島 266042)
馬來酸氟伏沙明是由雅培制藥研發的一種治療抑郁癥和強迫癥藥物,是全球第一種選擇性5-羥色胺再攝取抑制劑(SSRIs)類型的抗抑郁藥物,也是唯一一種單環類SSRIs[1-4]。與傳統的抗抑郁藥物相比,馬來酸氟伏沙明具有擬腎上腺素效能低,不易引起興奮、多汗等癥狀;抗膽堿和抗組胺效能低,很少引發體重增加和嗜睡等現象;藥物間相互作用小,有利于聯合用藥等優點。臨床統計表明,馬來酸氟伏沙明用藥時對胃腸道、中樞神經系統、性功能等方面具有一定副作用[5-7]。
氘代標記化合物是利用氘的同位素效應,將化合物中的氫原子或部分氫原子用氘原子替換的化合物,在生物醫學、藥物代謝動力學中都發揮著重要作用[8-10]。美國食品藥品監督管理局(FDA)頒布的工業指南要求在新藥研究中,藥物代謝安全評價必須使用同位素標記技術。將氘代技術應用到現有藥物的合成中,不會影響分子構象,有效性與安全性經過驗證,可用于改善早期開發藥物的部分缺點及副作用。本研究將馬來酸氟伏沙明中的甲基進行氘代化,合成穩定同位素標記的馬來酸氟伏沙明-D3,有助于進一步分析馬來酸氟伏沙明在人體內的代謝過程以及藥物動力學,為改善其副作用奠定基礎。
目前,國內穩定同位素標記藥物的研發和生產處于起步階段,市售的馬來酸氟伏沙明-D3為代理進口產品,價格昂貴,關于其合成技術還未見文獻報道,嚴重限制了該標準品在我國的廣泛使用。已報道的馬來酸氟伏沙明的合成方法主要有兩種:(1)以對三氟甲基苯腈為原料,與格氏試劑反應直接制備目標化合物[11-14]。該路線是目前文獻報道較多的方法,但反應周期長、重復性差,且使用高毒性氰類化合物為原料,使用時存在安全隱患。(2)以對三氟甲基苯甲酸為原料,在PCl3催化下,與甲氧基甲基胺反應生成Weinreb酰胺,進而與格氏試劑反應得到馬來酸氟伏沙明[15]。該反應過程用到PCl3,易造成環境污染,產生大量酸性廢液,增加了三廢處理成本,且不適合甲基氘代化。因此,探索一種穩定性高、簡潔高效合成馬來酸氟伏沙明-D3的方法是一項具有現實意義的工作。
本文首次以市售的4-芐氧基-1-丁醇(2)為原料,與氘代碘甲烷反應,得到氘代甲基化中間體(3),后續經過氫化脫芐、溴化、格氏反應、戴斯-馬丁氧化、縮合、取代、成鹽等反應,合成高純度馬來酸氟伏沙明-D3(1, Scheme 1),具體合成路線如Scheme 1所示。該化合物的合成方法現已申請專利保護,公開號為CN 113861066[16]。
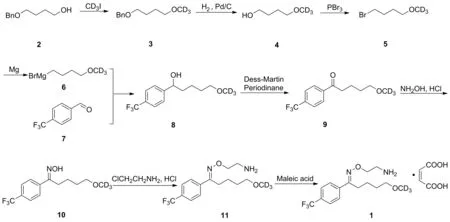
Scheme 1
1 實驗部分
1.1 儀器與試劑
Bruker AC-500 MHz型核磁共振波譜儀;Ultima Global spectrometer型高分辨質譜儀(德國英福康公司);WATERS-e2695型高效液相色譜儀(美國沃特斯)。
4-芐氧基-1-丁醇、三溴化磷、鹽酸羥胺、2-氯代乙基胺鹽酸鹽、馬來酸、碘、4-三氟甲基苯甲醛(99%,阿達瑪斯試劑有限公司);氘代碘甲烷(D: 99.9%,阿達瑪斯試劑有限公司);氫化鈉(60%,阿達瑪斯試劑有限公司);無水四氫呋喃(Water≤50 ppm,99.5%,阿達瑪斯試劑有限公司);Dess-Martin試劑(97%, Sigma-Aldrich);鎂屑(99%, Sigma-Aldrich);鈀碳催化劑(干基,10% Pd/C,阿拉丁試劑有限公司);其余所用試劑均為分析純,青島華東化學試劑有限公司。
1.2 合成
(1) ((4-(甲氧基-D3)丁氧基)甲基)苯(3)
500 mL三口瓶中,將4-芐氧基-1-丁醇(1, 18.0 g, 100 mmol)溶于N,N-二甲基甲酰胺(DMF, 200 mL),體系降溫至0~5 ℃,攪拌下加入氫化鈉(60%,分散于液狀石蠟)(6.0 g, 150 mmol),加畢,攪拌10 min;滴加氘代碘甲烷(29.0 g, 200 mmol),滴畢,反應體系自然升至室溫,攪拌反應12 h。體系倒入冰水中,加入乙酸乙酯(200 mL),分液,有機相依次用水(200 mL)、飽和食鹽水(200 mL)洗滌,無水硫酸鈉干燥,抽濾,濾液減壓濃縮得無色液體319.3 g,收率98%,不作純化直接下一步投料。
(2) 4-(甲氧基-D3)丁-1-醇(4)
在500 mL三口瓶中,將((4-(甲氧基-D3)丁氧基)甲基)苯(3, 17.7 g, 90 mmol)溶于無水甲醇(180 mL),加入鈀碳催化劑(1.77 g,鈀碳催化劑中鈀含量為10 wt%),于氫氣氣氛中室溫下快速攪拌12 h。停止反應,過濾,濾餅用無水甲醇(10 mL)洗滌,濾液減壓濃縮至恒重得4-(甲氧基-D3)丁-1-醇(4)9.5 g,收率98%,無色液體,不作純化直接下一步投料。
(3) 1-溴-4-(甲氧基-D3)丁烷(5)
在500 mL三口瓶中,氬氣保護,在大約0~5 ℃左右溫度下,將三溴化磷(24.6 g, 91 mmol)緩慢滴加至4-(甲氧基-D3)丁-1-醇(4, 7.5 g, 70 mmol)的四氫呋喃(140 mL)溶液中,加畢,自然升至室溫攪拌12 h。加入飽和碳酸氫鈉溶液調pH至7~8,乙酸乙酯(150 mL)萃取分液,有機相依次用水(100 mL)、飽和食鹽水(100 mL)洗滌,無水硫酸鈉干燥,抽濾,濾液減壓濃縮,柱層析分離得1-溴-4-(甲氧基-D3)丁烷(5)9.9 g,收率83%,無色液體。1H NMR(400 MHz, CDCl3)δ: 3.53~3.41(m, 4H), 2.05~1.91(m, 2H), 1.81~1.68(m, 2H);13C NMR(125 MHz, CDCl3)δ: 72.7, 65.2, 34.1, 29.8, 28.3。
(4) (4-(甲氧基-D3)丁基)溴化鎂(6)
氬氣保護下,250 mL三口瓶中加入鎂屑(1.44 g, 60 mmol),一小粒碘、無水四氫呋喃(30 mL),加熱至微沸,體系棕色褪去,滴加1-溴-4-(甲氧基-D3)丁烷(5, 8.5 g, 50 mmol)的無水四氫呋喃(30 mL)溶液,保持微沸狀態,加畢,攪拌1 h制得(4-(甲氧基-D3)丁基)溴化鎂(6)的四氫呋喃溶液。
(5) 5-(甲氧基-D3)-1-(4-(三氟甲基)苯基)-1-戊醇(8)
在氬氣保護下,0~5 ℃左右的溫度下,將4-三氟甲基苯甲醛(7, 8.7 g, 50 mmol)的無水四氫呋喃(20 mL)溶液滴加至上步制得的(4-(甲氧基-D3)丁基)溴化鎂(6)的四氫呋喃溶液中,加畢,自然升至升溫攪拌3 h,加入1 mol/L鹽酸(10 mL)淬滅反應,加入水(100 mL)、乙酸乙酯(100 mL)萃取分液,有機相用飽和食鹽水(100 mL)洗滌,無水硫酸鈉干燥,抽濾,濾液減壓濃縮,柱層析分離得5-(甲氧基-D3)-1-(4-(三氟甲基)苯基)-1-戊醇(8)11.1 g,兩步收率84%,白色固體;1H NMR(400 MHz, CDCl3)δ: 7.60(d,J=6.4 Hz, 2H), 7.46(d,J=6.4 Hz, 2H), 4.76(s, 1H), 3.36(t,J=5.1 Hz, 2H), 2.16(d,J=2.2 Hz, 1H), 1.81~1.71(m, 2H), 1.63(s, 1H), 1.62~1.58(m, 2H), 1.51~1.48(m, 1H), 1.41~1.36(m, 1H);13C NMR(125 MHz, CDCl3)δ: 147.8, 128.7, 125.1, 124.4, 124.3, 122.1, 72.7, 71,4, 37.9, 28.3, 21.3。
(6) 5-(甲氧基-D3)-1-(4-三氟甲基苯基)-1-戊酮(9)
500 mL反應瓶中加入5-(甲氧基-D3)-1-(4-(三氟甲基)苯基)-1-戊醇(8, 10.6 g, 40 mmol)、Dess-Martin試劑(25.4 g, 60 mmol)和二氯甲烷(100 mL),室溫攪拌2 h,加入飽和碳酸氫鈉溶液(100 mL),攪拌10 min,萃取分液,水相用二氯甲烷(100 mL)萃取,合并有機相,用飽和食鹽水(100 mL)洗滌,無水硫酸鈉干燥,抽濾,濾液減壓濃縮、干燥得5-(甲氧基-D3)-1-(4-三氟甲基苯基)-1-戊酮(9)10.1 g,收率96%,白色固體。1H NMR(400 MHz, CDCl3)δ: 8.05(d,J=6.5 Hz, 2H), 7.72(d,J=6.5 Hz, 2H), 3.42(t,J=5.1 Hz, 2H), 3.02(t,J=5.6 Hz, 2H), 1.85~1.80(m, 2H), 1.69~1.67(m, 2H);13C NMR(125 MHz, CDCl3)δ: 198.8, 139.4, 134.8, 128.1, 125.4, 125.3, 124.4, 72.1, 63.8, 38.3, 28.8, 20.6。
(7) (E)-5-(甲氧基-D3)-1-(4-(三氟甲基)苯基)-1-戊酮肟(10)
250 mL反應瓶中加入鹽酸羥胺(3.1 g, 45 mmol)、氫氧化鉀(5.1 g, 90 mmol)和無水乙醇(100 mL),室溫攪拌30 min,加入5-(甲氧基-D3)-1-(4-三氟甲基苯基)-1-戊酮(9, 7.9 g, 30 mmol),回流3 h。停止反應,降至室溫,加入水(100 mL)、二氯甲烷(100 mL)萃取分液,水相用二氯甲烷(50 mL)萃取,合并有機相,用飽和食鹽水(100 mL)洗滌,無水硫酸鈉干燥,抽濾,濾液減壓濃縮、柱層析得(E)-5-(甲氧基-D3)-1-(4-(三氟甲基)苯基)-1-戊酮肟(10)7.8 g,收率91%,白色固體。1H NMR(400 MHz, CDCl3)δ: 7.71(d,J=6.5 Hz, 2H), 7.53(d,J=6.5 Hz, 2H), 3.40(t,J=5.0 Hz, 2H), 2.68(t,J=5.6 Hz,2H), 1.75~1.67(m, 2H), 1.53~1.47(m, 2H);13C NMR(125 MHz, CDCl3)δ: 157.5, 134.1, 133.3, 127.6, 125.1, 124.9, 124.4, 72.3, 63.3, 36.9, 28.0, 19.9。
(8) (E)-5-(甲氧基-D3)-1-(4-三氟甲基苯基)-1-戊酮-O-(2-氨基乙基)肟(11)
250 mL反應瓶中加入(E)-5-(甲氧基-D3)-1-(4-(三氟甲基)苯基)-1-戊酮肟(10, 7.0 g, 25 mmol)、2-氯代乙基胺鹽酸鹽(2.9 g, 25 mmol)、氫氧化鉀(3.1 g, 55 mmol)和N,N-二甲基甲酰胺(80 mL),室溫攪拌6 h。停止反應,減壓濃縮,殘渣加入2 mol/L鹽酸調pH大約至1~2,加入水(100 mL)、二氯甲烷(100 mL)萃取分液,水相用10%氫氧化鈉水溶液調pH大約至11~13,用二氯甲烷(100 mL)萃取,有機相用無水硫酸鈉干燥,抽濾,濾液減壓濃縮至恒重得(E)-5-(甲氧基-D3)-1-(4-三氟甲基苯基)-1-戊酮-O-(2-氨基乙基)肟(11)7.8 g,收率92%,無色液體,不作純化直接下一步投料。
(9) 馬來酸氟伏沙明-D3(1)
250 mL反應瓶中加入(E)-5-(甲氧基-D3)-1-(4-三氟甲基苯基)-1-戊酮-O-(2-氨基乙基)肟(11, 6.4 g, 20 mmol)、馬來酸(2.3 g, 20 mmol)和無水乙醇(60 mL),室溫攪拌2 h,濾液減壓濃縮得馬來酸氟伏沙明-D3粗品,加入乙酸乙酯和正己烷的混合液(40 mL,其中乙酸乙酯和正己烷的體積比為1 ∶9),打漿1 h,抽濾,濾餅用正己烷(20 mL)洗滌,干燥得馬來酸氟伏沙明-D3(1) 8.4 g,收率96%,純度99.3%,白色固體。1H NMR(400 MHz, CDCl3)δ: 8.22(brs, 2H), 7.66(d,J=6.6 Hz, 2H), 7.56(d,J=6.6 Hz, 2H), 6.14(s, 2H), 4.39(t,J=3.5 Hz, 2H), 3.38~3.32(m, 4H), 2.73(t,J=5.7 Hz, 2H), 1.53(t,J=2.6 Hz, 4H);13C NMR(125 MHz, CDCl3)δ: 169.0, 158.7, 137.4, 134.8, 130.5, 130.2, 125.8, 124.5, 124.4, 124.0, 121.8, 70.9, 68.9, 38.9, 28.0, 25.2, 21.9; HR-MS(ESI)m/z: calcd for C15H19D3F3N2O2{[M+H]+}322.1822, found 322.1819。
1.3 結構表征
馬來酸氟伏沙明-D3的1H NMR譜圖如圖1所示。譜圖積分為18個氫(胺基上的兩個活潑氫未出峰),與天然豐度的馬來酸氟伏沙明相比,甲氧基上3個氫原子被氘取代,在高場區域未出現甲基峰,與馬來酸氟伏沙明-D3的結構吻合。
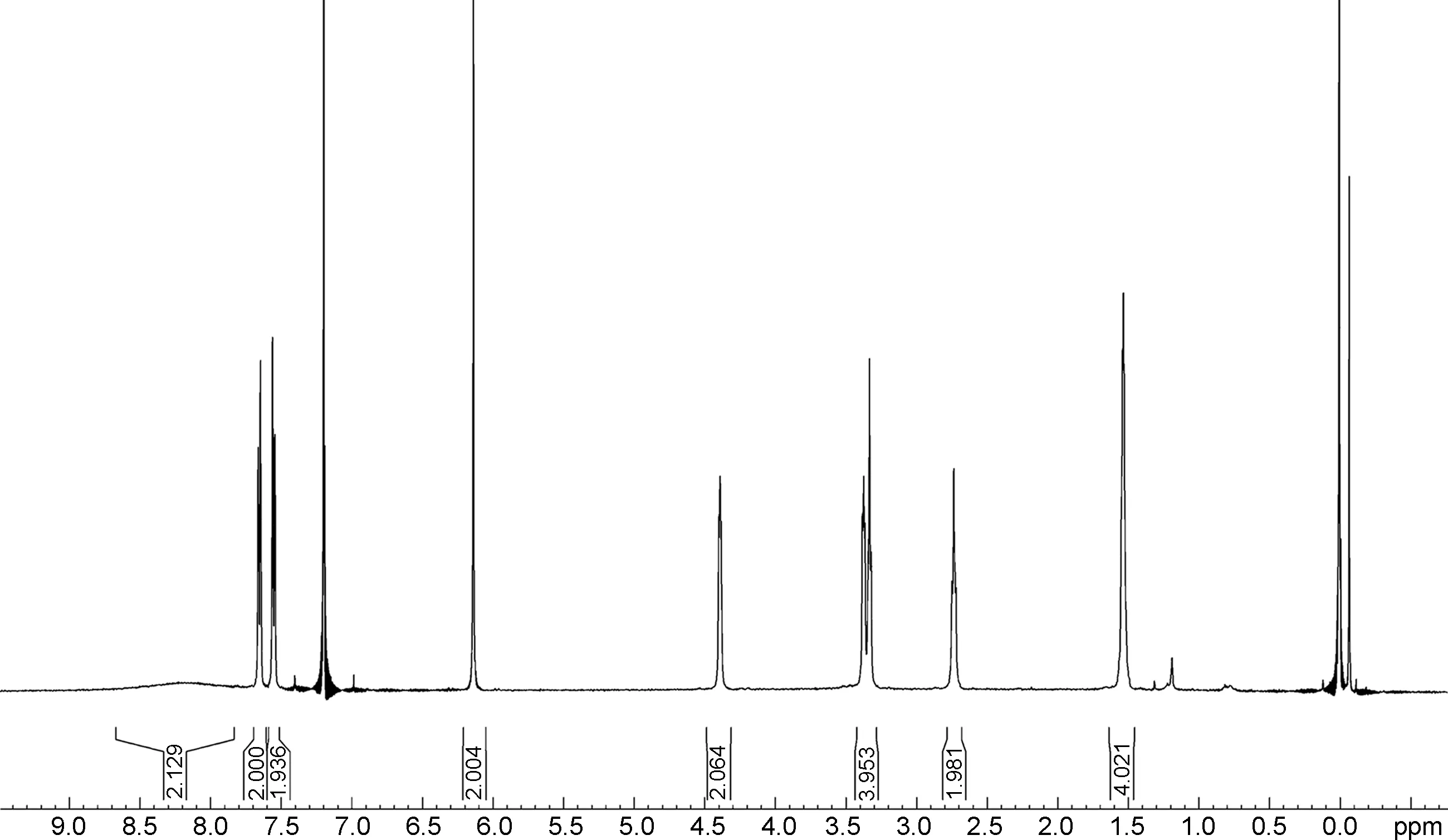
δ
馬來酸氟伏沙明-D3的高分辨質譜(圖2)中最高峰m/z322.1819為氟伏沙明-D3的加氫峰,即[M+H]+,其誤差為0.93 ppm,結合其氫譜數、據可以確定合成的產品為馬來酸氟伏沙明-D3。
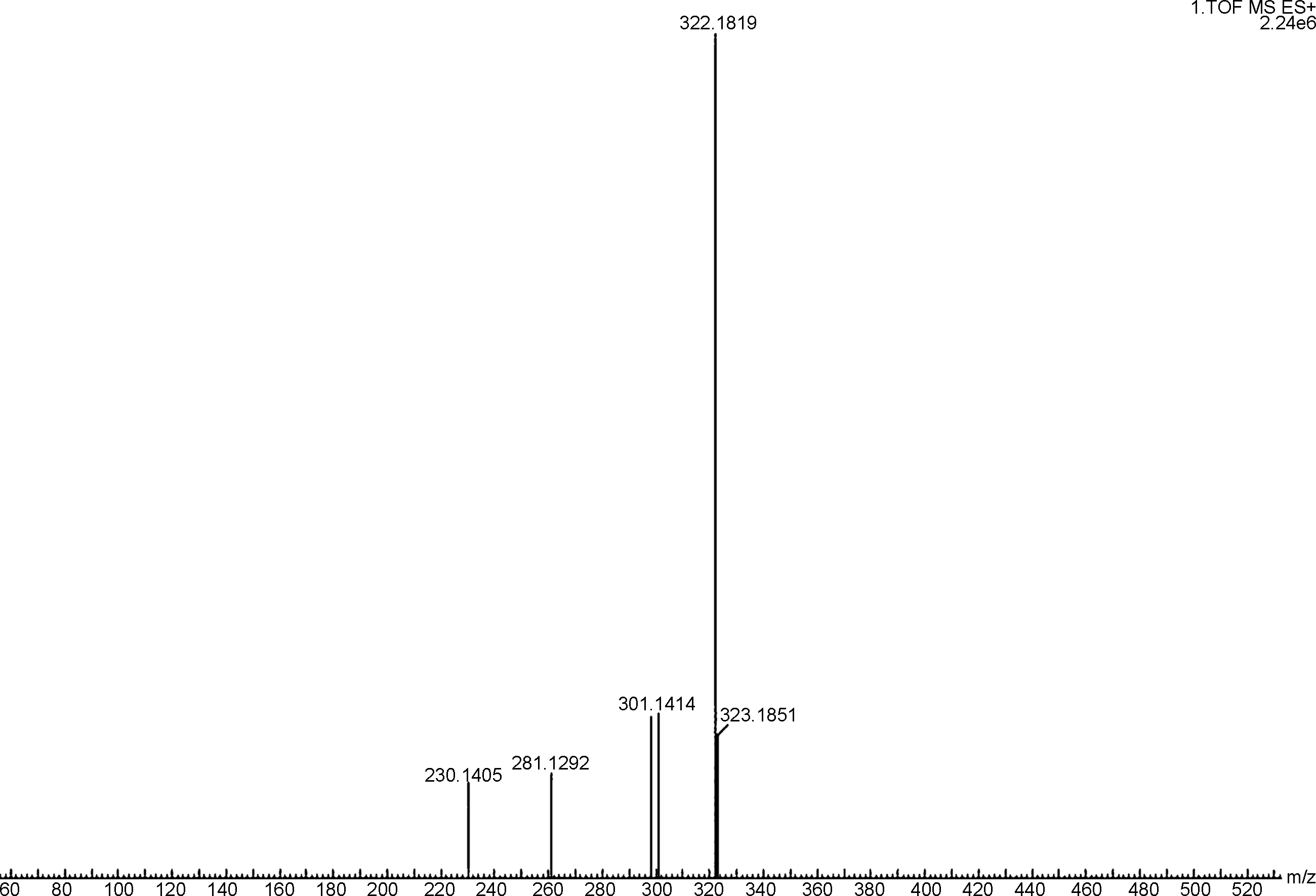
m/z
5-(甲氧基-D3)-1-(4-三氟甲基苯基)-1-戊酮(9)是合成馬來酸氟伏沙明-D3的關鍵中間體,由4-三氟甲基苯甲醛與格氏試劑(6)經親核加成、氧化反應制得。本文對反應過程中所涉及的溶劑溫度、投料量等因素進行了考察,以確定最優反應條件。
2.1 親核加成反應過程的優化
(1) 反應溶劑對格氏試劑制備的影響
格氏試劑的制備通常在非質子性溶劑中進行,以乙醚、四氫呋喃為反應溶劑進行考察,發現溶劑的不同對格氏試劑的生成具有直接影響。乙醚引發時間較長,如果體系無水環境控制不嚴格,會導致引發失敗。而四氫呋喃引發速度快,加入引發劑后,保持微沸狀態即可引發反應。同時,四氫呋喃的安全性更高,在工業化應用中比乙醚更具優勢,因此,將四氫呋喃作為該步反應的最佳溶劑。
(2) 反應溫度對親核加成反應的影響
在格氏試劑制備完成后,考察了反應溫度對親核加成反應的影響,結果見表1。由表1可知,當反應在回流狀態下進行時,收率僅有10%,說明溫度過高會導致副反應增加。隨著溫度的降低,收率相應提高,在0 ℃下反應收率可達87%,進一步降低溫度至-10 ℃,反應12 h收率出現一定下降,說明溫度過低會導致反應活性的下降。最終,確定0 ℃為反應的最佳溫度。
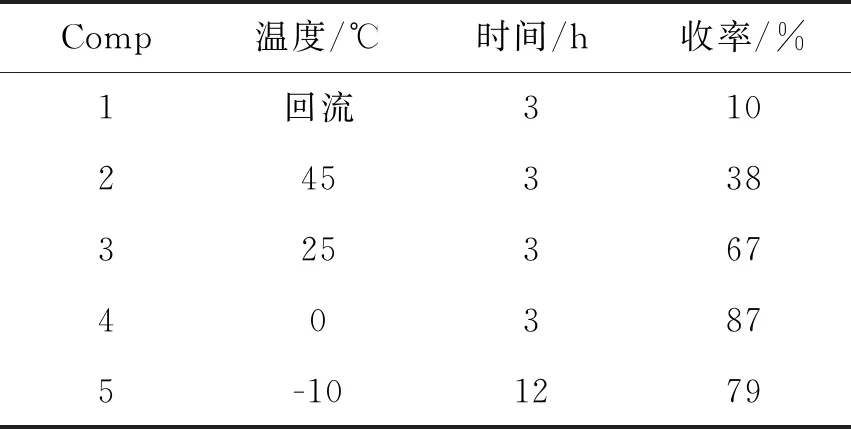
表1 溫度對反應的影響
2.2 氧化過程的優化
Dess-Martin氧化反應具有反應速率快、條件溫和、氧化劑用量少等特點,是現代有機合成中常用的氧化反應[17-18]。本研究首次將Dess-Martin氧化反應用于馬來酸氟伏沙明-D3關鍵中間體9的合成中,重點從氧化劑的用量、反應溶劑等工藝參數進行了條件優化。
(1) 氧化劑用量對反應的影響
在Dess-Martin氧化反應中,氧化劑用量對氧化性能有著至關重要的作用,如表2所示,以底物5-(甲氧基-D3)-1-(4-(三氟甲基)苯基)-1-戊醇(8)為基準,當使用Dess-Martin試劑為1.0當量時,反應活性較差,反應延長至12 h,收率也僅為76%。隨著氧化劑用量的增加,反應活性有明顯提高,當使用1.2當量氧化劑時,收率達到了最佳值。進一步增加用量,收率再無提高。因而,最終確定Dess-Martin試劑的最佳投料量為1.2當量。
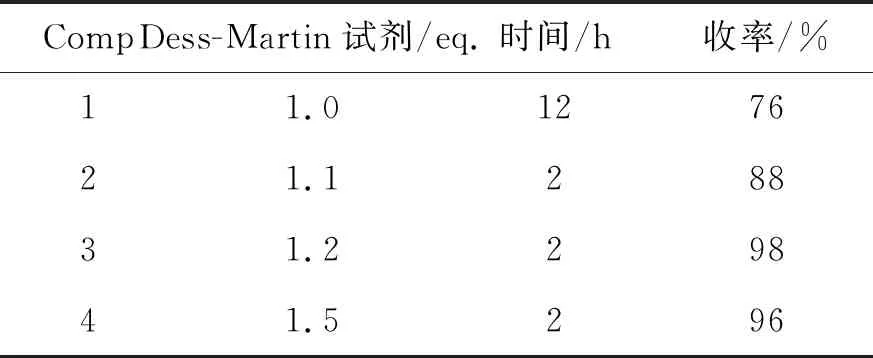
表2 氧化劑用量對反應的影響
(2) 反應溶劑對氧化反應的影響
在確定了Dess-Martin試劑的用量后,研究了溶劑對氧化反應的影響(表3)。從表3可以看出,該反應在非極性溶劑中能夠得到非常好的收率,其中鹵代類溶劑二氯甲烷效果最好,其次是四氫呋喃和甲苯;在極性溶劑乙酸乙酯和乙腈中反應也可以發生,但反應轉化率較差。最終將二氯甲烷作為該反應的優選溶劑。
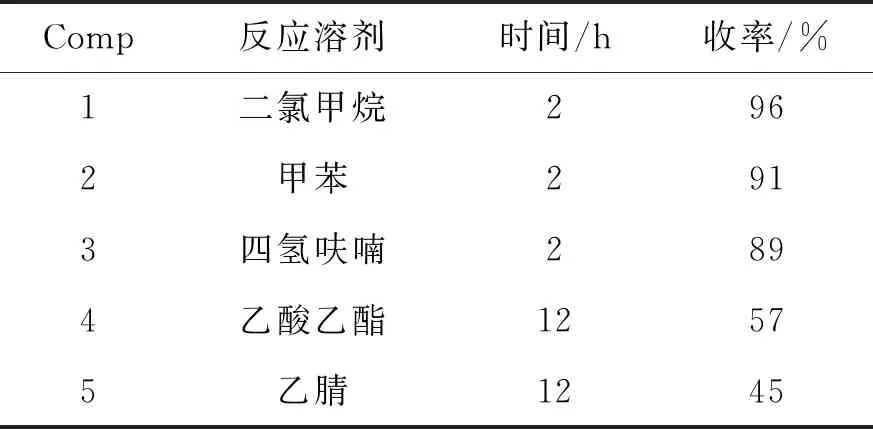
表3 溶劑對氧化反應的影響
以氘代碘甲烷為氘源,以4-芐氧基-1-丁醇(2)為起始原料,經甲基化、氫化脫芐、溴化、格氏反應、戴斯-馬丁氧化、縮合、取代、成鹽等反應,以51.7%總收率制備得到穩定的同位素標記的馬來酸氟伏沙明-D3。其中,5-(甲氧基-D3)-1-(4-三氟甲基苯基)-1-戊酮(9)的制備是合成馬來酸氟伏沙明-D3的關鍵步驟,本發明首次使用4-三氟甲基苯甲醛為原料與格氏試劑反應簡潔高效制備出5-(甲氧基-D3)-1-(4-(三氟甲基)苯基)-1-戊醇(8),經Dess-Martin氧化得到中間體9。相對于傳統方法,避免了反應周期長、重復性差及使用高毒性氰類化合物等問題。馬來酸氟伏沙明-D3作為內標藥物使用,可應用于臨床藥代動力學方面研究,從而更準確和方便地了解馬來酸氟伏沙明在人體內的代謝過程和作用機制。
