生血方的鑒別與含量測定
2022-09-14 11:54:18郭懷宇陳金鋒謝俊杰郁冬冬
藥學與臨床研究 2022年4期
郭懷宇,陳金鋒,謝俊杰,郁冬冬,姜 蕾
六安市中醫院 藥劑科,安徽六安 237001
生血方是六安市中醫院生產的醫院制劑穴位貼,處方由黃芪、女貞子、當歸、炒白術、茯苓、菟絲子、枸杞子等10 味中藥組成,具有補腎、活血的功效。該方聯合他藥常規治療宮頸癌和胃癌等疾病,經臨床驗證,臨床效果理想[1,2]。黃芪中含有皂苷、黃酮、氨基酸等成分。其中,皂苷類成分具有抗癌等藥理作用[3,4];黃酮類成分具有抗缺血和改善血相作用[5,6]。女貞子中含有三萜、黃酮、多糖類成分,具有抗腫瘤、調節免疫等作用[7]。當歸中含有揮發油和有機酸等成分,具有補血、促進免疫等作用[8]。馬靖、孟楣等采用高效液相-蒸發光散射檢測器(HPLC-ELSD)測定黃芪中的黃酮類成分和皂苷類成分[9,10]。本研究通過黃芪等薄層色譜鑒別以及高效液相色譜法(HPLCELSD),建立了黃芪甲甘和芒柄花素的含量測定,用于控制該制劑的質量。
1 儀器與藥品、試劑
1.1 儀器
Agilent 1200 高效液相色譜儀(含奧泰ELSD 6000 蒸發光檢測器、CSChormPlus 工作站),美國安捷倫公司;BT125D 型電子分析天平(賽多利斯科學儀器北京有限公司);KQ-5200DA 型超聲波清洗器(昆山市超聲儀器有限公司);硅膠G 薄層板(青島海洋化工有限公司)。
1.2 藥品與試劑
黃芪甲苷對照品(批號:110781-201717)、芒柄花素對照品(批號:111703-201504),女貞子對照藥材(批號:121041-201404)、阿魏酸對照品(批號:121137-201606)均購于中國食品藥品檢定研究院;生血方(3 批:批號:20180521、20180618、20180713),六安市中醫院制劑室自制。乙腈為色譜純;其他試劑為分析醇;水為超純水。
2 方法與結果
2.1 黃芪薄層色譜鑒別
2.1.1 供試品溶液制備 取本品6g,研細,加甲醇100mL,超聲處理30 分鐘,濾過,濾液蒸干,固體物加水20mL 使溶解,用水飽和正丁醇提取4 次(40mL/次),合并正丁醇液,用氨試液洗滌2 次(40 mL/次),棄去氨試液,正丁醇液水浴蒸干,固體物加適量甲醇溶解,定容至1 mL,即得。
2.1.2 對照品溶液制備 取黃芪甲苷對照品適量,加甲醇制成每毫升含1 mg 的溶液,即得。
2.1.3 陰性對照溶液制備 取缺黃芪藥材,根據制劑工藝制成相應陰性對照,按“2.1.1”項下方法制備,即得。
2.1.4 鑒別方法 吸取供試品、對照品,陰性對照溶液各5 μL,點于同一硅膠G 薄層板上,以三氯甲烷-甲醇-水(13∶7∶2)的下層溶液(10 ℃以下分層靜置30 分鐘以上)為展開劑,展開,取出,晾干,噴以10%硫酸乙醇溶液,在105 ℃加熱約5 分鐘,結果見圖1A。由圖1A 可知,供試品色譜與對照品色譜相應的位置顯相同顏色斑點,陰性無干擾。
2.2 女貞子薄層色譜鑒別
2.2.1 供試品溶液的制備 取本品粉末2 g,加稀乙醇50 mL,超聲處理30 分鐘,濾過,濾液即為供試品溶液。
2.2.2 對照藥材溶液的制備 取女貞子對照藥材2g,同法制成對照藥材溶液。
2.2.3 陰性對照溶液制備 取缺女貞子各藥材,根據制劑工藝制成相應陰性對照,按“2.2.1”項下方法制備陰性對照溶液。
2.2.4 鑒別方法 吸取供試品、對照藥材、陰性對照溶液各5 μL,分別點于同一硅膠G 薄層板上,以石油醚(30 ℃~60 ℃)-乙酸乙酯(1∶1)為展開劑,展開,取出,晾干,置碘蒸氣中熏至斑點清晰,置日光下檢視,結果見圖1B。由圖1B 可知,供試品色譜與對照藥材色譜相應的位置顯相同顏色斑點,陰性無干擾。
2.3 當歸的薄層鑒別
2.3.1 供試品溶液的制備 取本品粉末6 g,加1%碳酸氫鈉溶液50mL,超聲處理10 分鐘,離心,取上清液用稀鹽酸調節pH 值至2~3,用乙醚振搖提取2 次,每次20mL,合并乙醚液,揮干,固體物加甲醇1mL 使溶解,即得。
2.3.2 對照品溶液的制備 取阿魏酸對照品適量,加甲醇制成每毫升含1 mg 的溶液,即得。
2.3.3 陰性對照溶液制備 取缺當歸各藥材,根據制劑工藝制成相應陰性對照,按“2.3.1”項下方法制備陰性對照溶液。
2.3.4 鑒別方法 吸取供試品溶液、對照品溶液、陰性對照溶液各10 μL,分別點于同一硅膠G 薄層板上,以環己烷-二氯甲烷-乙酸乙酯-甲酸(1∶1∶1∶0.1)為展開劑,展開,取出,晾干,置紫外光燈(365 nm)下檢視,結果見圖1C。由圖1C 可知,供試品色譜與對照品色譜相應的位置顯相同顏色斑點,陰性無干擾。

圖1 生血方中的薄層鑒別
2.4 黃芪甲苷和芒柄花素含量測定
2.4.1 溶液制備
2.4.1.1 對照品溶液 精密稱取黃芪甲苷、芒柄花素對照品適量,加甲醇制成每毫升含黃芪甲苷1.326 mg、芒柄花素0.894 mg 的的混合對照品溶液,搖勻即得。
2.4.1.2 供試品溶液 取生血方粉末約3 g,精密稱定,置具塞錐形瓶,加2%氫氧化鉀甲醇溶液100 mL,超聲處理(功率100 W,頻率40 kHz)30 分鐘,濾過,濾液蒸干,固體物加水10 mL 使溶解,用水飽和的正丁醇提取4 次(30 mL/次),合并正丁醇液,用氨試液洗滌2 次(3 mL/次),棄去氨試液,正丁醇液蒸干,固體物加水5 mL 溶解,過D101 型大孔吸附樹脂柱(內徑1.5 cm,長12 cm),以50 mL 水洗脫,棄去洗脫液,再用40%乙醇30 mL 洗脫,棄去洗脫液,繼用70%乙醇80 mL 洗脫,收集洗脫液,蒸干,固體物加甲醇溶解并轉移至5 mL 量瓶中,加甲醇稀釋至刻度,搖勻,即得。
2.4.1.3 陰性對照溶液 取缺黃芪陰性樣品,按照“2.4.1.2”項下方法制備缺黃芪的陰性對照溶液。
2.4.2 色譜條件與系統適用性試驗 色譜柱為Agilent ZORBAX SB-C18(4.6mm×250mm,5μm),流動相為乙腈-水(34:66),流速為1mL·min-1,柱溫為30 ℃;蒸發光散射檢測器漂移管溫度為100 ℃,載氣為空氣,載氣流量為2.6 L·min-1,進樣量為20 μL。
在上述色譜條件下,黃芪甲苷和芒柄花素的色譜峰分離比較理想,分離度>1.5,理論塔板數以黃芪甲苷和芒柄花素計算分別為14166 和23835,且陰性無干擾。對照品、樣品及陰性樣品色譜圖見圖2。
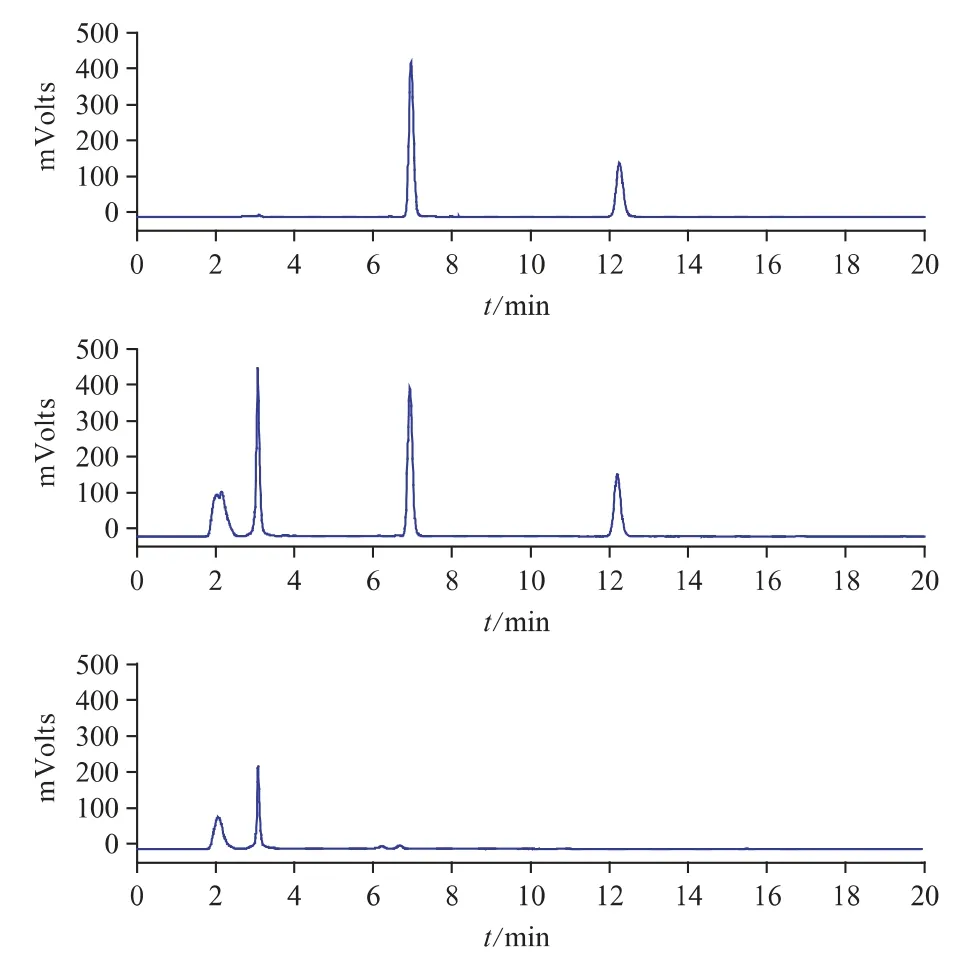
圖2 對照品(A)、樣品(B)、黃芪陰性樣品(C)的HPLC 色譜圖
2.4.3 線性關系考察 精密吸取混合對照品溶液0.2、0.4、0.8、1.2、1.6、2.0 mL,置于5 mL 量瓶中,加甲醇稀釋至刻度,搖勻,按“2.4.2”項下色譜條件進樣20 μL 測定。以進樣量的對數和對應峰面積對數得到回歸方程,結果見表1。

表1 回歸方程和線性范圍
2.4.4 進樣精密度試驗 取同一混合對照品溶液,連續進樣6 次,計算峰面積,結果黃芪甲苷和芒柄花素峰面積的RSD 分別為1.6%和1.4%。表明儀器精密度良好。
2.4.5 穩定性試驗 取同一供試品溶液,分別于0、2、4、8、12、24 h 進樣測定,測得黃芪甲苷和芒柄花素峰面積的RSD 分別為0.2%和0.9%,表明樣品溶液24 h 內穩定性較好。
2.4.6 耐用性試驗 選用3 種不同品牌的色譜柱[色譜柱1:Agilent Zorbax SB C18色譜柱(4.6 mm×250 mm,5 μm);色譜柱2:依利特Hypersil ODS2 色譜柱(4.6 mm×250 mm,5 μm),色譜柱3:Unitary C8色譜柱(4.6 mm×250 mm,5 μm)],分別取“2.4.1.2”項下的供試品溶液,按“2.4.2”項下的色譜條件進樣檢測,均能達到系統適用性試驗要求。
2.4.7 重復性試驗 取同一批號生血方樣品適量,共6 份,分別按“2.4.1.2”項下方法制備供試品溶液,連續進樣,記錄黃芪甲苷和芒柄花素峰面積,根據外標法,計算6 份樣品中各自所含的黃芪甲苷和芒柄花素的含量,測得黃芪甲苷和芒柄花素的含量分別為0.683 mg·g-1和0.477 mg·g-1,RSD 分別為0.3%和1.1%。表明該方法重復性良好。
2.4.8 加樣回收率試驗 取已知含量的生血方9份,每份1.5g,精密稱定,按“2.4.1.2”項下方法制備供試液,精密加入低、中、高混合對照品溶液,平行制備9 份樣品,按“2.4.2”項下色譜條件進樣測定,記錄黃芪甲苷和芒柄花素的峰面積,根據外標法計算平均加樣回收率,結果見表2。

表2 回收率試驗結果(n=9)
黃芪甲苷和芒柄花素的回收率分別在97.7%~100.1%和97.7%~98.8%,其平均回收率分別為99.1%和98.5%,RSD 分別為0.7%和0.3%。表明該方法的回收率符合要求。
2.4.9 樣品含量測定 分別取3 個批號生血方樣品適量,按“2.4.1.2”項下方法制備供試品溶液,按“2.4.2”項下色譜條件進樣測定,記錄黃芪甲苷和芒柄花素的峰面積,由回歸方程計算樣品中黃芪甲苷和芒柄花素的含量,結果見表3。
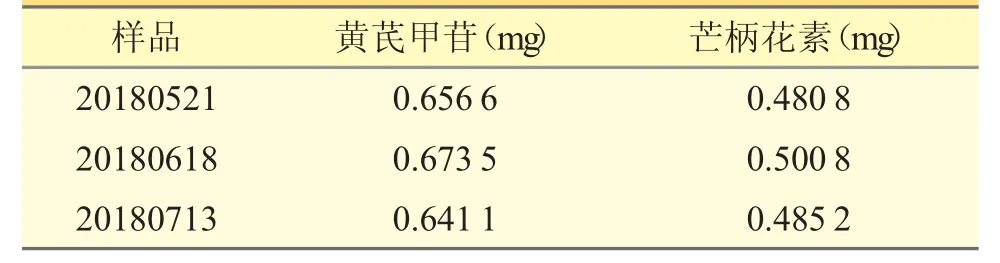
表3 樣品含量測定結果
3 討論
本試驗分別對甲醇/水,乙腈/水兩個流動相系統進行考察,得出乙腈/水為系統的流動相分離效果較為理想,供試品溶液中2 個成分有較好的峰形且能完全分離,溶劑噪音水平較低,所得圖譜較理想。
生血方是由10 味中藥組成,本實驗除了對生血方中黃芪、女貞子、當歸進行定性鑒定,以及對黃芪甲苷、芒柄花素2 種成分進行定量測定外,還針對黃精、山藥等其余7 種中藥進行了薄層鑒別,嘗試不同極性展開系統和顯色劑的效用,均未能獲得滿意結果,存在未見相同特征斑點、陰性樣品干擾等問題,故未將該部分納入質控研究中。
黃芪中的活性成分黃芪甲苷為皂苷類,芒柄花素為黃酮類,本試驗通過建立HPLC-ELSD 法同時檢測2 個成分含量的方法,為控制生血方質量提供了試驗基礎。
