一例兩性離子型Cu(Ⅱ) 配合物的合成及其結構研究*
2022-07-31 04:26:18王玉娜郭津榕馬鈺璐
云南化工 2022年7期
王玉娜,郭津榕,馬鈺璐**
(1.昆明醫科大學 藥學院暨天然藥物藥理重點實驗室,云南 昆明 65050;2.云南民族大學 化學與環境學院,云南 昆明 650504)
近年來,兩性離子型配合物由于具有電荷分離、水穩定性等優勢,使其在光致發光、主客體傳感、磁性等領域備受關注[1-6]。合成兩性離子配合物已經成為眾多化學家研究的重點。在合成過程中,有機配體的選擇對配合物的結構和性質起著關鍵作用[7-10]。通過引入離子型配體來構筑兩性離子配合物是一種較為常見,且易成功的方法。在兩性離子類配體中,吡啶鎓多羧酸兩性離子半剛性配體是一種自身優勢較高的配體,它兼具了常見兩性離子配體和普通羧酸配體的優點[11-15]:1)與傳統配體相比,兩性離子配體的正電荷和負電荷是分離的,所以它們具有很好的水溶性[16-21];2)它可以像多羧酸配體一樣擁有不同的程度的去質子化。利于合成出構型各異的配合物;3)與純剛性配體相比,它存在著易于旋轉的 -CH2- 基團,在配位過程中容易與金屬離子連接,形成不同維度的結構。
本文選用吡啶鎓多羧酸半剛性兩性離子有機化合物 1,1-((2,3,5,6-四甲基-1,4-亞苯基)雙(亞甲基))雙(4-羧基吡啶-1-鎓)二氯化物作為配體,在水熱條件下,配體通過羧酸基團的橋連作用與Cu2+結合,構筑了配合物{Cu0.5(HLCl2)H2O}。借助X-單晶衍射儀和CrystalExplorer 3.1軟件探究了配合物的形成過程和結構特點。
1 實驗部分
1.1 試劑與測試儀器
有機配體根據文獻報道進行合成,其中合成原料1,4-雙(氯甲基)-2,3,5,6-二甲苯和異煙酸均購買于探索平臺。Cu(NO3)2·6H2O和N,N-二甲基乙酰胺(DMF)購買于北京伊諾凱公司。去離子水由實驗室水處理器制取。單晶衍射數據來自于Bruker Smart APEX CCD 單晶衍射儀(MoKα= 0.705 mm-1)。霍夫曼表面分析由CrystalExplorer 3.1軟件完成。
1.2 合成有機配體1,1-((2,3,5,6-四甲基-1,4-亞苯基)雙(亞甲基))雙(4-羧基吡啶-1-鎓)二氯化物(H2LCl2)
稱取1,4-雙(氯甲基)-2,3,5,6-二甲苯(20 mmol,4.62 g)和異煙酸(50 mmol, 6.16 g)于 250 mL 的圓底燒瓶中,以 100 mL 的DMF作為反應溶液,加熱回流 72 h。然后將反應液進行冰浴,冷卻后有白色固體析出。通過過濾,將白色固體取出,并用DMF洗滌三次。洗滌完成后,在常溫下進行自然干燥,最終所得到的白色粉末即為所要合成的有機配體(H2LCl2)(產率為85.34%,以1,4-雙 (氯甲基)-2,3,5,6-四甲基苯為標準進行計算)。合成過程如圖1所示。
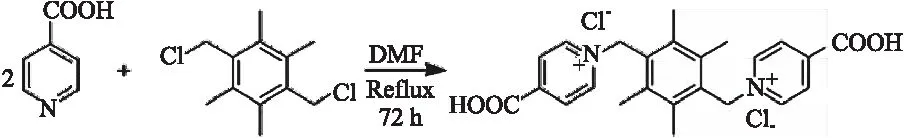
圖1 配體(H2LCl2)的合成過程
1.3 合成配合物{Cu0.5(HLCl2)H2O}
稱取有機配體H2LCl2(0.05 mmol,23.85 mg)和NaOH顆粒(0.10 mmol, 4 mg)于 10 mL 西林瓶中,加入 4 mL 去離子水,超聲 5 min 使配體完全溶解,此時溶液的pH值為7~8。然后加入Cu(NO3)2·6H2O(0.20 mmol,59.11 mg),再次超聲 15 min。超聲完成后,將蓋子旋緊,并放入 100 ℃ 的烘箱內保持 48 h,然后以 10 ℃/h 的速度降至室溫,并在室溫下放置 12 h,最后獲得藍色晶體(產率為62.51%,以有機配體H2LCl2為標準進行計算)。
2 晶體結構與表征
2.1 晶體測試和解析
在顯微鏡下,挑選出大小合適且無任何裂痕的晶體用于結構測試。在室溫下,用Bruker APEX-II CCD 單晶衍射儀進行衍射數據的采集。由SADABS程序包進行衍射數據的吸收校正[22]。在晶體解析方面,利用OLEX2軟件里的直接法對晶體骨架進行解析。通過ShelXT程序進行骨架中非氫原子的指認,同時利用XL程序里的全矩陣最小二乘法完成結構中非氫原子的精修,氫原子通過理論計算獲得。配合物的氫鍵數據來源于與OLEX2軟件相關聯的PLATON軟件。詳細的晶體學數據、主要的鍵長、鍵角數據和氫鍵數據見表1、表2和表3中。其中,CCDC號為217523。
2.2 配合物{Cu0.5(HLCl2)H2O}的晶體結構描述
配合物{Cu0.5(HLCl2)H2O}是一個零維分子,結晶在triclinic晶系P-1空間群。在它的不對稱單元中,包含半個二價Cu2+離子,一個脫去一個質子的配體分子(HLCl2)和一個配位水分子,如圖2所示。中心金屬Cu2+離子位于一個四邊形構型中,分別與來自配位水分子的兩個氧原子(O2、O21)和來自不同配體分子的兩個羧基氧原子(O1、O11)配位(圖2a)。Cu—O鍵的鍵長分別是0.1912(2) nm和0.1937(2) nm,O—Cu—O的鍵角范圍是88.77°(11)~180.0°。羧基采取單齒橋連的 μ1-η1η0配位模式與金屬離子相連。由于—CH2—基團的旋轉作用,使配體兩端的苯環與中間苯環處于不同的平面,配體呈現出一個“凹”字形。最后,通過C—H···O和O—H···O兩種氫鍵作用(C—H···O氫鍵來自苯環上的氫原子與羧基上的氧原子、O—H···O氫鍵來自配位水分子上的氫原子與羧基上的氧原子),相鄰的零維分子間可以堆積成如圖2b所示的三維超分子結構。
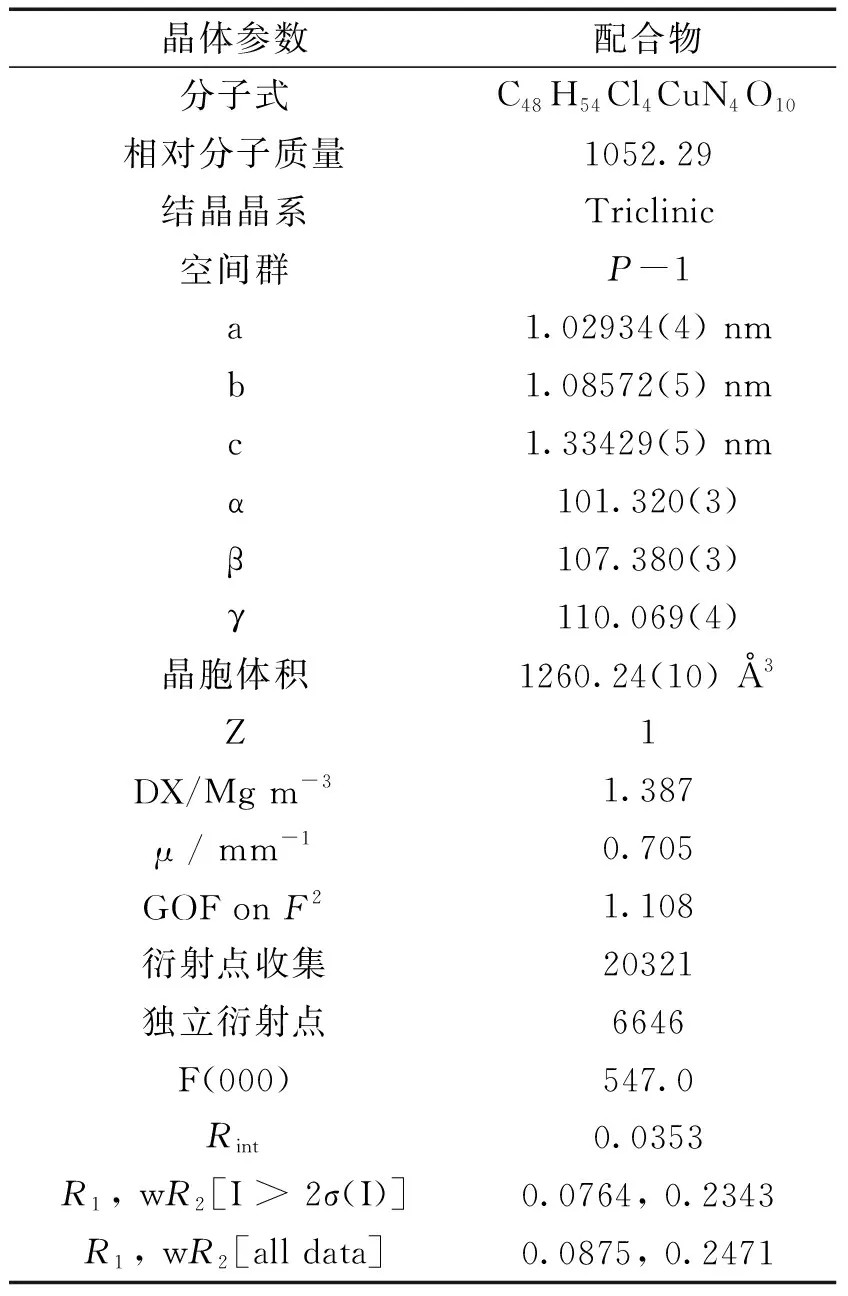
表1 配合物的晶體學參數和精修結果
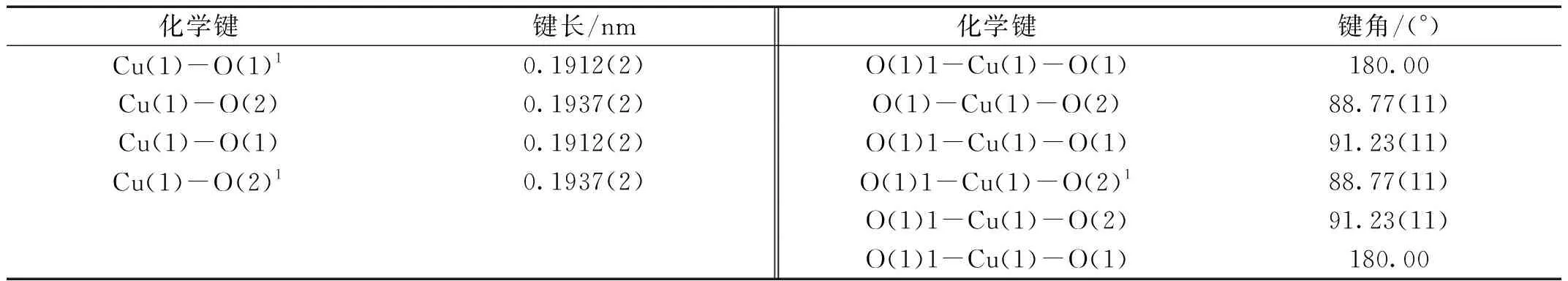
表2 配合物部分的鍵長和鍵角

表3 配合物中氫鍵的鍵長和鍵角
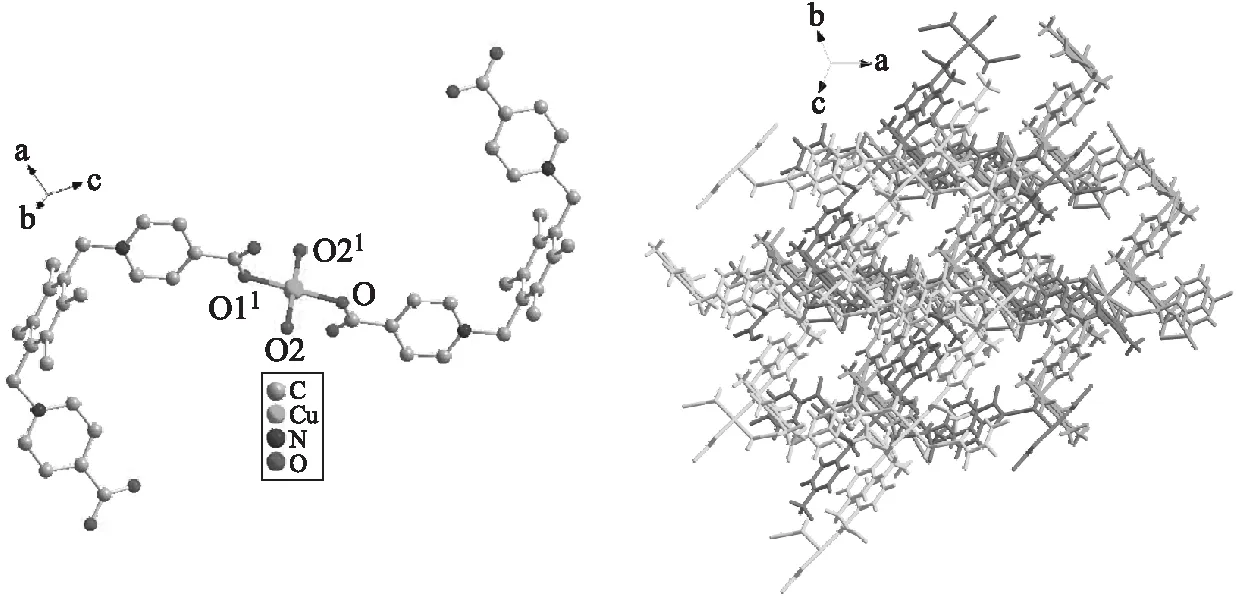
(a) (b)
3 配合物{Cu0.5(HLCl2)H2O}晶體結構的霍夫曼表面分析
為了更好的了解三維超分子結構中零維分子間的相互作用和堆積模式,我們利用 CrystalExplorer 3.1軟件對配合物進行了霍夫曼表面分析。在霍夫曼表面的dnorm圖譜中,晶體結構表面的紅點代表著在這個位置與相鄰的分子間存在著強相互作用力,藍色代表著相互作用力沒有或者很弱,而灰色代表著中等強度的作用力,如圖3所示。配合物表面的紅點位置主要是羧基和金屬中心的位置,根據分析是氫鍵作用和配位作用引起的。從二維指紋圖譜兩端的尖峰可以知道,氫鍵作用主要來自于配位水分子上的氫原子和配體羧基上的氧原子形成的O···H氫鍵。為了從定量的角度來確定各種相互作用力,我們進行二維指紋圖譜的分析。從圖3中看出,在配合物中H···H/H···H、Cl···H/H···Cl和O···H/H···O相互作用力占比較大,分別為30.5%、20.2% 和19.9%,而相對較少的C···H/H···C和C···O/O···C相互作用力,他們分別占總相互作用力的 11.2% 和2.0% 。
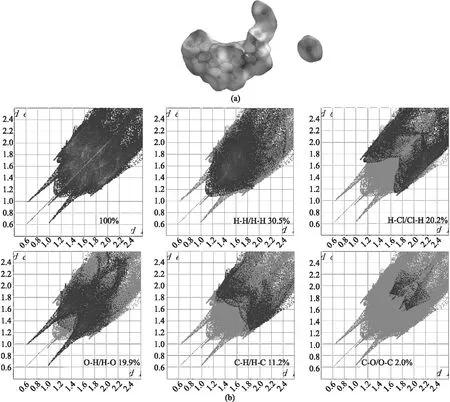
圖3 晶體結構的霍夫曼dnorm圖譜和二維指紋圖譜
4 結語
本文利用吡啶鎓多羧酸半剛性兩性離子配體 1,1-((2,3,5,6-四甲基-1,4-亞苯基)雙(亞甲基))雙(4-羧基吡啶-1-鎓)二氯化物與金屬鹽Cu(NO3)2在水熱條件下進行自組裝,構筑了配合物{Cu0.5(HLCl2)H2O}。結構分析表明,該配合物是一個典型的零維分子,通過分子間氫鍵作用,最終可拓展成一個三維超分子結構。經霍夫曼表面分析研究表明,配合物中存在H···H/H···H、Cl···H/H···Cl、O···H/H···O、C···H/H···C和C···O/O···C等多種弱相互作用力,其占比分別為30.5%、20.2%、19.9%、11.2% 和2.0% 。
