左側額葉Rosai-Dorfman病1例
2021-11-06 03:25:20姚國杰
臨床神經外科雜志 2021年10期
關鍵詞:癲癇
曹 杰 韋 可 姚國杰
1 病例資料
60 歲男性,因突發意識喪失2 h 入院。體格檢查未見明顯神經系統陽性體征。入院頭顱CT示左側額部可見一稍高密度影,周邊可見水腫帶(圖1A)。頭顱MRI 增強示左側額頂部顱板下見一寬基底、半圓形高信號影,大小約25 mm×28 mm×19 mm,邊界清楚,鄰近腦實質受壓,并可見無強化水腫信號(圖1B~D)。腦電圖可見異常慢波。術前診斷左側額葉腦膜瘤。神經導航及電生理監測下開顱手術,術中見腫瘤呈灰紅色,質地較韌,血供豐富,與周圍組織界限尚清,術中血管保護良好、未損傷靜脈。術后病理診斷為左側額葉Rosai-Dorfman 病。術后復查頭部CT 顯示術區水腫緩解緩慢(圖1E~G),脫水治療時間較長。復查24小時長程視頻腦電圖可見異常慢波。術后未予放化療。出院時術區仍有水腫。術后隨訪6個月未再發癲癇,偶有頭暈,無其他不適。術后6個月復查頭顱MRI增強示水腫消失(圖1H~I)。
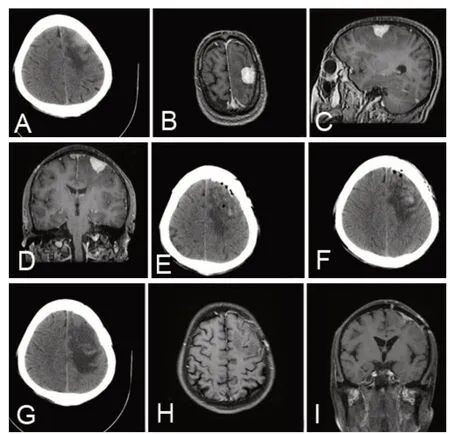
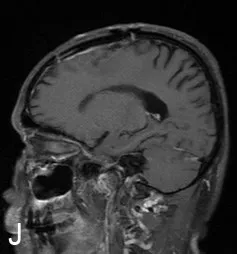
圖1 左側額葉Rosai-Dorfman 病手術前后影像學表現
2 討論
羅道病(Rosai-Dorfman disease,RDD)又稱竇組織細胞增生伴巨淋巴結病,是一種罕見的、病因不明的良性組織細胞增生性疾病,病因尚不明確,可能和病毒感染、免疫系統失調有關。RDD 可累及全身各部位,最常見于淋巴結,也可出現淋巴結外受累,如鼻腔、口腔、咽喉、眼眶、軟組織、皮膚、骨骼、下呼吸系統、泌尿生殖系統等。顱內RDD極為少見。
原發性顱內RDD的臨床特征和影像學表現無特異性,臨床表現多與病變的位置、大小及壓迫程度有關,可表現為頭痛、癲癇、肢體癱瘓、感覺異常、垂體功能紊亂、神經功能障礙等為主,部分病人可出現顱內壓增高癥狀。原發性顱內RDD的確診依靠病理檢查。該病需與朗格漢斯組織細胞增生癥、富于淋巴漿細胞型腦膜瘤、惡性的纖維組織細胞瘤、慢性非特異性炎癥等相鑒別。
由于顱內RDD罕見,治療方案及效果尚不明確。目前認為手術切除病變是顱內RDD最有效的治療方法,多數病人術后無復發。本文病例腫瘤全切除術后復查腦電圖可見異常慢波,出院后未口服抗癲癇藥物;隨訪6個月無癲癇發作,復查MRI增強示水腫消失、腫瘤未復發。
總之,顱內RDD非常罕見,影像學表現易誤診為腦膜瘤,因此,對腦膜或腦表面有類似影像學表現時,除考慮腦膜瘤外,還需考慮顱內RDD;以癲癇為單發癥狀顱內原發RDD,若腫瘤完全切除,即使術后復查腦電圖異常慢波,也不需要預防性應用抗癲癇藥物;術后顱內水腫可能存在較長時間,甘露醇及白蛋白治療效果欠佳,若無頭痛、反應遲鈍、意識障礙等顱內壓增高癥狀,可臨床觀察;顱內RDD 沒有規范的治療指南,即使是目前認為最有效的完全切除病變,也存在復發,因此長期隨訪對治療方案及評估預后具有重要意義。
猜你喜歡
中國民間療法(2021年5期)2021-06-09 09:21:04
中華養生保健(2020年2期)2020-11-16 00:49:00
解放軍醫學院學報(2020年12期)2020-03-29 05:11:46
中成藥(2017年6期)2017-06-13 07:30:35
飲食科學(2017年5期)2017-05-20 17:11:53
臨床醫藥文獻雜志(電子版)(2017年11期)2017-05-17 04:48:10
安徽醫科大學學報(2015年9期)2015-12-16 11:09:44
中國當代醫藥(2015年7期)2015-03-01 02:01:13
西南軍醫(2015年4期)2015-01-23 01:19:30
西部中醫藥(2014年6期)2014-03-11 16:07:47
