瑞德西韋及其合成中間體結構和性質的計算分析
2021-10-16 01:48:20陳蒙儀孫祥黃羅儀王朝杰
溫州醫科大學學報 2021年9期
陳蒙儀,孫祥,黃羅儀,王朝杰
1.溫州醫科大學附屬第一醫院 藥學部,浙江 溫州 325015;2.溫州醫科大學 藥學院,浙江 溫州 325035
瑞德西韋是一種由美國Gilead生物技術公司所生產的核糖核苷類似物,主要干擾病毒RNA轉錄過程,具有廣譜抗病毒活性[1]。早期臨床研究報道了瑞德西韋首次用于治療蘇格蘭女性護士埃博拉腦膜炎,并且完成了治療埃博拉病毒感染的三期臨床試驗[2]。在前期細胞和動物實驗中,瑞德西韋均顯示出對嚴重急性呼吸綜合征冠狀病毒(severe acute respiratory syndrome coronavirus,SARSCoV)和中東呼吸綜合征冠狀病毒(middle east respiratory syndrome coronavirus,MERS-CoV)有較好的抗病毒活性。小鼠實驗證明瑞德西韋預防和治療性用藥均可改善肺功能,降低肺病毒載量,減輕肺病理學損傷[3-4]。這也是在新型冠狀病毒肺炎(corona virus disease 2019,COVID-19)暴發后選擇瑞德西韋進行臨床試驗的原因之一。HOLSHUE 等[5]首次報道了一位美國COVID-19患者在使用瑞德西韋后臨床癥狀迅速減輕的情況,此后關于瑞德西韋治療COVID-19的研究大量開展。因此,瑞德西韋及其合成中間體的結構和性質是值得探索的,SIEGEL等[6]報道了該藥的合成工作,涉及兩代合成路線,見圖1。
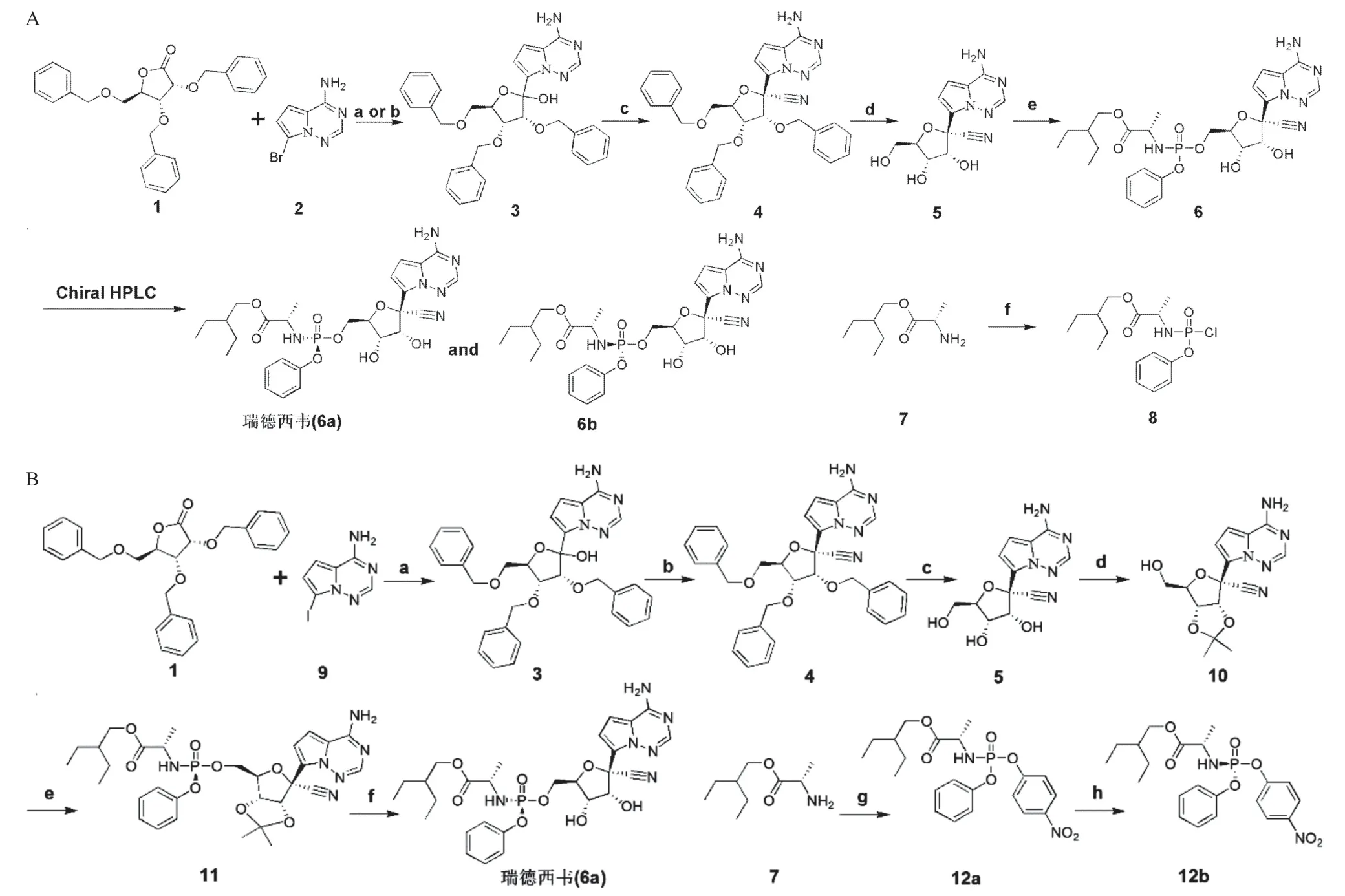
圖1 瑞德西韋兩代合成路線
從反應合成路線可知,瑞德西韋(6a)是由鳥嘌呤類似物和戊糖組成核苷,再通過磷酸修飾得到的。瑞德西韋是治療COVID-19的潛在候選藥物,然而,關于瑞德西韋的結構性質及其合成中間體的反應活性位點等方面的理論研究甚少。本研究將通過量子化學方法和藥代動力學計算,對瑞德西韋及其合成中間體13種化合物的分子結構、反應活性位點、紅外光譜等進行分析,并利用ACD/Percept平臺預測這13種化合物的藥代動力學參數并對其進行成藥性評價。
1 計算方法
采用密度泛函理論方法X3LYP,在6-311+G(2d,p)基組水平上對瑞德西韋及其合成中間體的幾何結構進行優化,然后進行紅外光譜計算,得到無虛頻的能量極小結構,得到波函數進行電子結構分析。有研究報道X3LYP雜化密度泛函方法的頻率一致性較好[7],KOSTJUKOVA等[8]用密度泛函理論計算研究吖啶橙染料在水溶液中的振動吸收光譜,分析了使用各種泛函和基組的計算,結果表明X3LYP方法的理論值與實驗值最吻合。密度泛函計算利用Gaussian 16[9]程序實現,GaussView 6.0用于結構可視化,通過Multiwfn3.6[10]程序,對分子表面靜電勢、原子電荷、前線分子軌道進行分析,預測活性反應位點,并基于概念密度泛函活性指數(化學勢、化學硬度、親電指數、親核力差值指數以及親電力差值指數)重點對化合物5和6a進行分析。
2 結果和討論
2.1 幾何結構分析 所有化合物均在X3LYP水平上用6-311+G(2d,p)進行了結構優化,相應的理論計算幾何結構均為穩定結構。據報道[6],在瑞德西韋以及其合成中間體中,瑞德西韋(6a)和中間體(12b)均得到了單晶結構,因此將理論計算參數和晶體數據進行比較。6a和12b的優化幾何構型和原子編號如圖2所示,表1列出了它們的部分結構參數(鍵長和鍵角)和XRD單晶衍射得到的結構參數,對實驗數據和計算數據進行線性回歸,得到6a和12b的鍵長和鍵角計算值與實驗值的線性擬合圖。以化合物6a的鍵長擬合為例,得到計算值與實驗值的函數關系為:y=1.00639x-0.00773,其中y是6a的鍵長計算值,x是實驗值,R2=0.981,接近于1,表明實驗值和計算值吻合得很好;此外,化合物6a的鍵角擬合函數關系為y=0.99373x+1.02514,R2=0.969;化合物12b的鍵長擬合函數關系為y=1.04285x-0.06025,R2=0.994,鍵角擬合函數關系為y=1.01187x-0.67776,R2=0.980,都接近于1,體現理論計算值與實驗值吻合得很好,也說明選取的計算方法是可靠的。

表1 瑞德西韋(6a)和中間體(12b)的計算及實驗的部分鍵長(?)和鍵角(°)
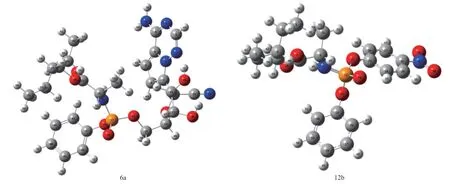
圖2 優化后的瑞德西韋(6a)和中間體(12b)的分子結構及原子編號
2.2 反應活性位點
2.2.1 分子表面靜電勢(electrostatic surface potential,ESP):ESP是通過識別親核和親電攻擊的潛在位點來表征分子反應活性位點的一種方法。ESP常被用于研究分子間的相互作用、反應部位以及分子識別,可以解釋分子化學反應活性與分子性質(局部電荷、偶極距、電負性等)之間的關系,ESP越負的位置對應的原子越容易發生親電反應,越正的位置對應的原子越容易發生親核反應[11]。優化13種分子結構后分析其表面靜電勢,ESP圖上顏色深淺反映靜電勢的強弱,靜電勢為負的區域用藍色表示,易受到親電試劑的進攻;靜電勢為正的區域用紅色表示,易受到親核試劑的進攻。分子表面不同區域有若干極大值點和極小值點,其中,橙色小球對應靜電勢極大值點,青色小球對應靜電勢極小值點,見圖3。我們發現靜電勢在全局的最大值點大都位于吡咯并三嗪環上的氨基氫或支鏈胺基氫附近(除化合物1),表明此區域親核性較高,容易發生親核反應。靜電勢在全局的最小值點大都位于羥基氧或芐氧基的氧原子附近,這是因為O的電負性比較強,周圍電子云密度較大,具有親電活性,易發生親電反應。因此推斷戊糖環上的芐氧基或羥基氧易受到親電試劑的攻擊,而鳥嘌呤類似物環上的氨基氫或支鏈胺基氫易受到親核試劑的攻擊。這與SIEGEL等[6]研究結果相一致。
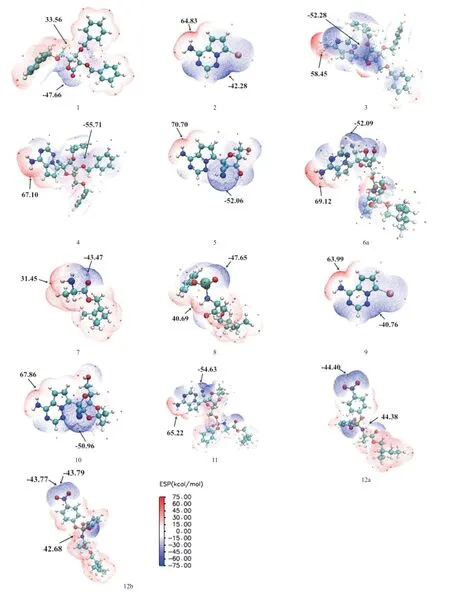
圖3 13種化合物在X3LYP/6-311+G(2d,p)水平下的靜電勢等值面圖
2.2.2 原子電荷:原子偶極矩校正的Hirshfeld電荷可表征分子內不同原子所帶電荷大小,是探究分子親核或親電特性強弱及確定化學活性位點的一種有效方法,其中帶負電荷越多的原子相應的親電活性可能也越強[12]。計算得到瑞德西韋及其具相同母核五種中間體的Hirshfeld電荷,對于化合物3,其中羥基氧O5(-0.72 a.u.)帶有最多的負電荷,戊糖環氧O4(-0.41 a.u.)次之,表明羥基氧O5具有較好的親電活性,羥基氫H45(0.49 a.u.)和氨基氫H72(0.45 a.u.)、H73(0.47 a.u.)都具有較高正電荷,表明這幾個位置親核活性較高,易發生親核反應。同理對于化合物4,帶負電荷順序是戊糖環氧O4(-0.44 a.u.)>三個芐氧基O5(-0.39 a.u.)、O6(-0.34 a.u.)、O8(-0.34 a.u.),氨基氫H72(0.46 a.u.)、H73(0.47 a.u.)具有較高正電荷;化合物5,負電荷是三個羥基氧O5(-0.70 a.u.)、O6(-0.74 a.u.)、O8(-0.71 a.u.)多于戊糖環氧O4(-0.50 a.u.),羥基氫H25(0.45 a.u.)、H26(0.48 a.u.)、H29(0.44 a.u.)和氨基氫H33(0.48 a.u.)、H34(0.49 a.u.)都具有較高正電荷;化合物6a,帶負電荷順序是羥基氧O35(-0.71 a.u.)、O36(-0.76 a.u.)>磷氧基O18(-0.69 a.u.)>羰基氧O25(-0.62 a.u.)>戊糖環氧O11(-0.44 a.u.)>芐氧基O19(-0.38 a.u.),羥基氫H71(0.47 a.u.)、H72(0.47 a.u.),氨基氫H69(0.48 a.u.)、H70(0.48 a.u.)和支鏈胺上氫H51(0.44 a.u.)都具有較高正電荷;化合物10與11有類似的分布情況。可以看出瑞德西韋及其中間體結構上羥基氧附近是具有最多的負電荷分布,最易發生親電反應,氨基氫和羥基氫附近親核活性較高,易發生親核反應。這與分子表面靜電勢得到的結果相一致。有研究表明,N-羥乙酰神經氨酸分子的羥基氧原子具有多的負電荷,有較強的親電活 性[13];高子飛等[14]的研究結果表明綠原酸分子中的羥基氫原子所帶正電荷較大;賈壯等[15]用量子化學方法指導普萘洛爾類似物的合成研究中,通過原子電荷分布和靜電勢圖確定反應位點,這些都與本研究的結論和依據相似。
2.2.3 前線軌道分析:根據前線分子軌道理論可知,前線分子軌道在分子反應過程中具有重要作 用[16]。前線軌道包括最高占據軌道(highest occupied molecular orbital,HOMO)和最低未占軌道(lowest unoccupied molecular orbital,LUMO),HOMO電子優先提供,而LUMO也先接受電子。HOMO和LUMO之間的能隙是分子化學穩定性的指標,也是決定分子電荷流動特性的重要參數,能隙(Δε(LUMO-HOMO))越小,表示分子中電子越容易發生躍遷,反應活性就越強,能隙越大,電子被激發的難度也越大,分子的穩定性也會較好。嚴茜茜等[17]研究表明通過HOMO與LUMO的能極差能反應化合物活性大小。前線軌道分布圖上的藍色區域代表軌道波函數負相位,綠色區域代表軌道波函數正相位。從前線軌道分布圖可看出,只要含有吡咯并三嗪環結構的化合物,HOMO和LUMO軌道都集中分布在吡咯并三嗪環上,說明瑞德西韋及其具相同母核中間體的吡咯并三嗪環上的原子是主要的親電或親核反應位點,即鳥嘌呤類似物結構是瑞德西韋的母核結構,是發揮抑制作用的活性中心。這與金興輝 等[18]用量子化學研究1,2,3-三氨基胍二硝基胍鹽中,因大部分HOMO和LUMO分布在二硝基胍陰離子上,說明二硝基胍陰離子是該化合物的反應活性位點的依據相似。所有化合物的HOMO和LUMO軌道能量均為負值,說明這些化合物的電子結構比較穩定,且化合物5的Δε(LUMO-HOMO)相對比6a的小,說明化合物5具有較高的活性。
2.3 紅外吸收光譜 紅外光譜是分子中基團由于發生伸縮振動、變形振動等產生的,其可用于定性分析分子結構。13種化合物在800~1 800 cm-1處都有不同強度的吸收,見圖5。在1 100 cm-1附近有CO-C的伸縮振動峰,歸屬于化合物的芐氧基和戊糖環結構;在1 500 cm-1附近都有強吸收峰,歸屬于吡咯并三嗪環及苯環的C=N和C=C的伸縮振動;在1 600 cm-1附近有N-H彎曲振動引起的較強吸收峰,對應吡咯并三嗪環的氨基。其他特征吸收峰如在1 200 cm-1附近的峰,歸屬于6a、8、11、12a、12b的P=O伸縮振動;在1 750 cm-1附近有C=O伸縮振動引起的強吸收峰,對應1、6a、7、8、11、12a、12b的酯基C=O結構;化合物4、5、6a、10、11都在2 300 cm-1附近有吸收峰,是由氰基C≡N的伸縮振動引起的;在3 500 cm-1和3 800 cm-1附近的O-H吸收峰,歸屬于化合物3、5、6a、10中羥基的O-H伸縮振動。對比化合物1、3、4、5的譜圖,可以看出三個芐氧基的C-O-C的吸收峰,變成三個羥基O-H的吸收峰;對比化合物5、10、11的譜圖,可以看出從化合物5有3處O-H吸收峰,到化合物10只有1處O-H吸收峰,到化合物11無O-H吸收峰。根據對計算所得的譜圖信息進行分析歸屬,與理論的頻率范圍基本吻合。并且我們也可以看出化合物的羥基氧、芐氧基、羥基氫、胺基氫均是活性反應位點,與前面分子靜電勢和原子電荷得到的結論一致。文獻[6]中沒有提供各合成中間體或產物的紅外光譜數據。
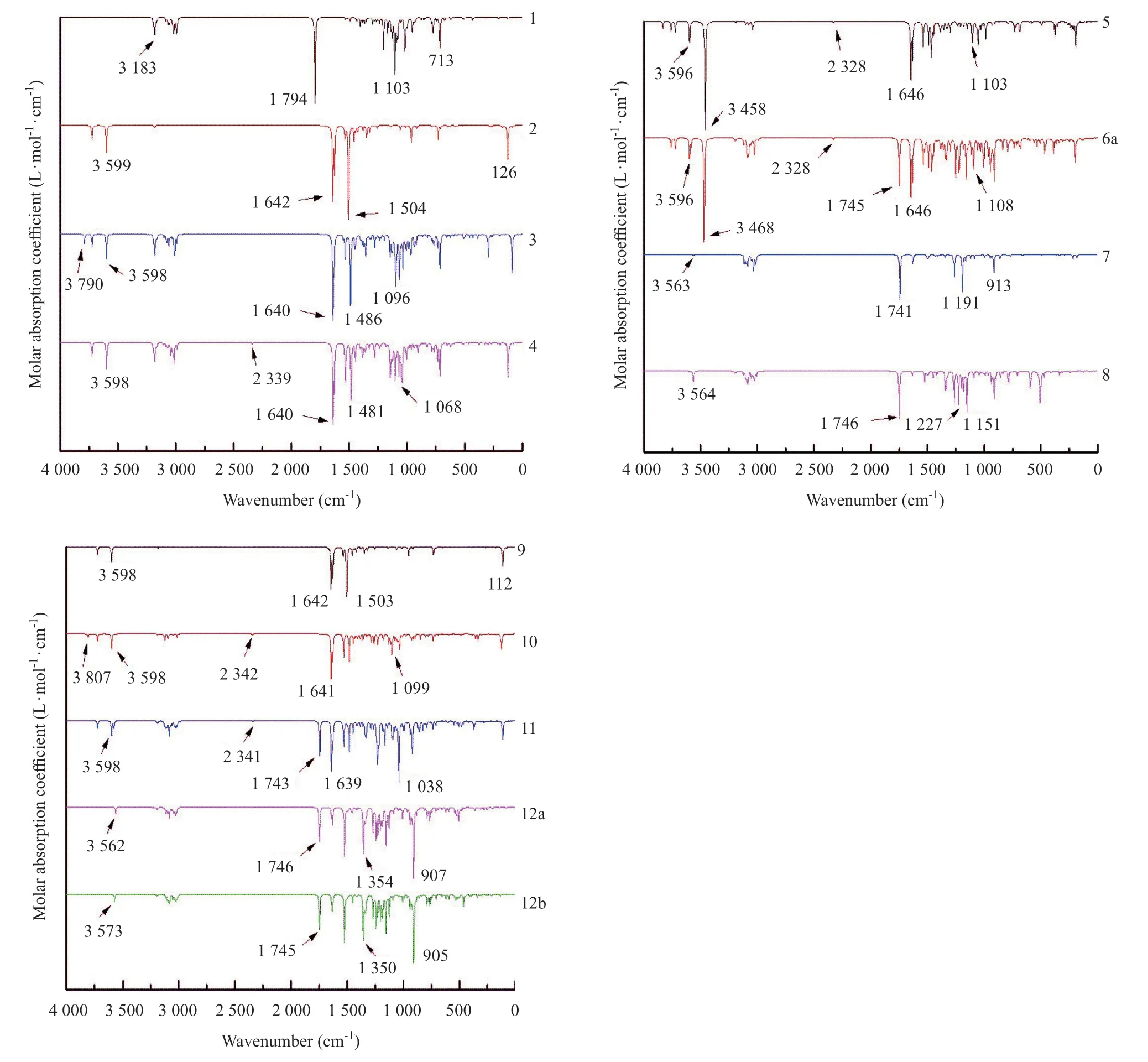
圖5 13種化合物在X3LYP/6-311+G(2d,p)水平下的紅外譜圖
2.4 綜合比較中間體(5)和瑞德西韋(6a)的藥理活性作用 YAN等[19]研究表示母體核苷5是瑞德西韋到達肺部的主要代謝物,由于其合成的簡單性和在獸醫環境中的體內功效,認為5在COVID-19治療中優于瑞德西韋(6a)。我們通過密度泛函理論方法,分析中間體(5)和瑞德西韋(6a)的藥理活性與分子結構之間的關系,為后續藥效關系的深入研究提供理論基礎。
從表2中可知,化合物5的O4、O5、O6、O8、C11、C13、N14等原子具有較大的負電荷,說明這些原子易與受體蛋白的正電荷區域結合而發揮作用,對比瑞德西韋(6a)中相應結構的原子電荷,發現化合物5原子上所帶的電荷稍多,更易與受體蛋白的正電荷區域作用。因此,從電荷分析可以預測,化合物5與受體蛋白的作用強于瑞德西韋(6a),即化合物5可能具有較強的藥理活性。
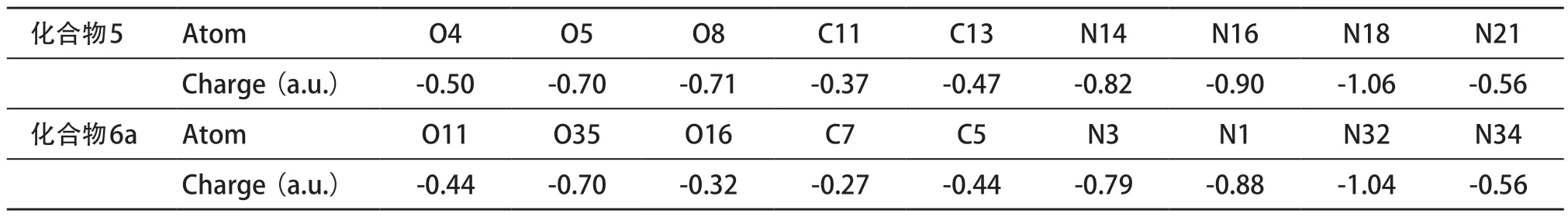
表2 化合物5和6a的部分原子電荷
根據上述圖4 的前線軌道分布圖,化合物5和6a的HOMO和LUMO軌道基本相近,均集中分布在吡咯并三嗪環上。由它們的能級差可知,化合物5的 Δε(LUMO-HOMO)略小于6a,表明其穩定性較弱,活性較6a強。SIEGEL等[6]研究的瑞德西韋的合成方法中說到,經過對化合物5及其結構相似的化合物進行細胞實驗,發現化合物5比其他化合物對病毒活性作用較強,說明化合物5的活性和選擇性在篩選的化合物中都呈現一定優勢。
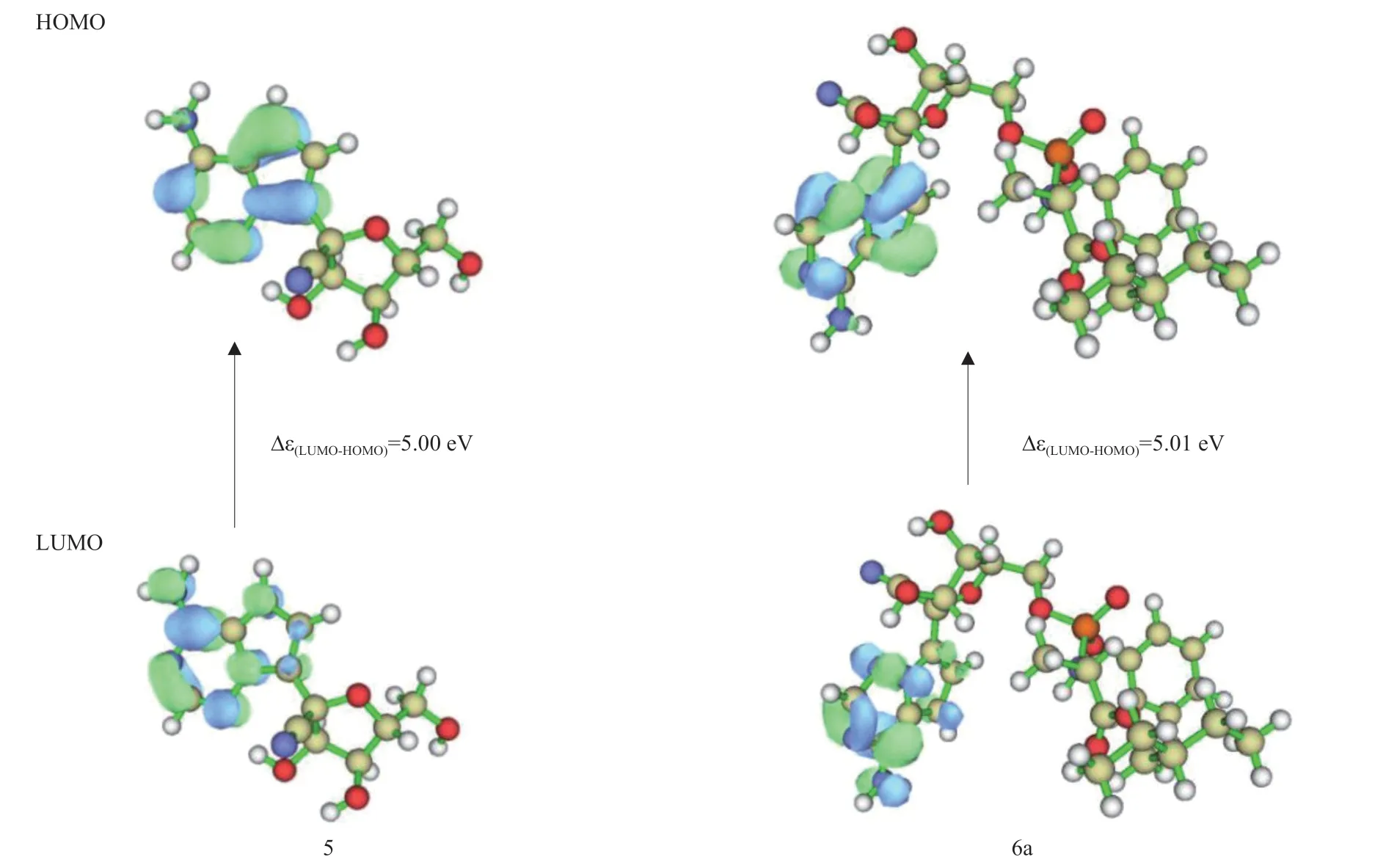
圖4 化合物5和6a在X3LYP/6-311+G(2d,p)水平下的HOMO和LUMO軌道分布圖及能差
概念密度泛函活性指數(如化學勢μ、化學硬度η、親電指數ω、親核力差值指數Δεn、親電力差值指數Δεe等)常被應用于化合物活性研究。化學勢μ越小,化學硬度η越大,分子越穩定,反之分子越不穩定,活性越大。親電指數ω表示分子對親電試劑的結合程度,而親核力差值指數Δεn和親電力差值指數Δεe則表示分子的親核和親電活性大小[20-21]。根據化合物5 和6a的概念密度泛函活性指數,化合物6a的化學勢μ(-4.019 eV)比5 的化學勢μ(-4.012 eV)低,6a的化學硬度η(4.624 eV)比5的化學硬度η(4.621 eV)大,即6a的穩定性比5好,則化合物5的活性較大;化合物6a的親電指數ω(1.746 eV)比5的親電指數ω(1.742 eV)大,即6a對親電試劑結合程度比5大;化合物5的親核力差值指數Δεn(0.040 eV)比6a的親核力差值指數Δεn(0.040 eV)大,親電力差值指數Δεe(8.065 eV)比6a的親電力差值指數Δεe(8.077 eV)小,即化合物5親核的活潑性能較大,親電的活潑性能比6a小。
綜上所述,化合物5的活性比瑞德西韋(6a)強,我們推測化合物5 的治療作用可能優于瑞德西韋(6a),需待進一步開展更多的體外和動物實驗去驗證。
2.5 藥代動力學分析 ACD/Percepta是基于QSAR模型精確計算化合物的吸收(absorption,A)、分布(distribution,D)、代謝(metabolism,M)、排泄(excretion,E)、毒性(toxicity,Tox)以及理化(PhysChem)等方面性質的新一代預測化合物藥代動力學參數的軟件。ADME/Tox特性在藥物發現領域中發揮著重要作用,計算ADME/Tox特性評估的目的是正確預測候選藥物的體內藥代動力學特性。有研究表明,60%的藥物因不良ADME/Tox性質而退出臨床試驗[22]。因此在新藥研發早期開展候選藥物的成藥性研究有利于縮短藥物的研發周期,降低研發成本,同時提高新藥開發的成功率。在這項研究中,主要關注的是評價理化性質,如氫鍵供體數(hydrogen bond donor,HBD)、氫鍵受體數(hydrogen bond acceptor,HBA)、拓撲極性表面積(topological polar surface area,TPSA)、親脂性(lipophilicity,LogP)、水溶性(water solubility,LogS)、藥代動力學,如胃腸吸收、BBB通透劑、P-gp底物和藥物相似性。除此之外,還考慮了一些重要的毒性參數,如急性口服毒性、致癌性、基因毒性[23]。
藥物通過細胞膜的滲透性直接受到藥物分子量和拓撲極性表面積(topological polar surface area,TPSA)的影響。分子量較高、TPSA較高的藥物不利于吸收。LogP代表親脂性,是指藥物在有機相和水相中分配系數的對數,此性質影響藥物的吸收,LogP值較高,藥物越易吸收。LogS與溶解度相關,可接受的溶解度范圍為不溶<-10<難溶<-8<微溶<-6<可溶<-2<易溶<0。HBD和HBA影響藥物穿過細胞膜的能力。可旋轉鍵的數量代表口服生物利用度,應該保持在10以下[24-25]。
Lipinski類藥五規則(rule-of-five,Ro5)是廣泛用于評估化合物類藥性是否良好的經驗規則,類藥性的篩選有利于獲得藥動學藥效學性質平衡的候選藥物[24]。符合Lipinski規則的化合物可能有更好的藥代動力學性質,在生物體內代謝過程中有更高的生物利用度,因而也更有可能成為口服藥物。13種化合物的理化性質預測結果見表3。化合物2、5、7、8、9、10、12a、12b均符合Lipinski五規則,類藥性較好。
13 種化合物的藥代動力學參數見表4。其中Caco-2細胞源于人體結腸癌細胞,結構和功能與腸上皮近似,細胞內有藥物代謝酶。Caco-2細胞模型是一種研究藥物轉運機制和預測藥物腸吸收的體外篩選工具[26],從藥物吸收的預測結果來看,除化合物5,其余吸收性均較好。血漿蛋白結合率(plasma protein binding,PPB)即當藥物進入人體后與血漿內白蛋白結合的比例。藥物在血漿中常以結合型和游離型兩種形式存在,只有游離型藥物才具有藥物活性,所以PPB對藥物的治療效果有著重要影 響[27]。從藥物分布的預測結果來看,化合物1、3、4與藥物血漿蛋白結合能力較強;化合物2、9、12a、12b與藥物血漿蛋白結合能力中等。藥物代謝又稱為藥物的生物轉化,是機體對外源物質的處理方式。肝臟作為人體最重要的代謝器官,主要以細胞色素P450(CYP450酶)介導參與口服藥物的首過效應,其中CYP1A2、CYP2C9、CYP2C19、CYP2D6和CYP3A4參與了90%外源物的代謝活動[28]。表4中結果表明13種化合物對五種CYP450亞型的抑制作用都較低或沒有抑制作用,因此對體內代謝產生的影響較小。糖蛋白(P-gp)是一種外排轉運蛋白,細胞膜中的P-gp使藥物能夠在細胞內移動,對P-gp的抑制可能會干擾藥物的轉運[29]。從表中看出,沒有化合物是磷-糖蛋白底物的抑制劑。污染物致突變性檢測(Ames試驗)與癌癥有關,陽性的Ames試驗表明有可能導致突變,表中數據表明,13種化合物的毒性均不明確。
綜合表3和表4的參數,表明化合物2、9、12a、12b的藥代動力學評價較佳,然而前面重點討論的化合物5和6a的理化性質和藥代動力學的參數都不理想,我們推測瑞德西韋(6a)可能對COVID-19療效甚微或基本無效。SHRESTHA等[30]通過隨機對照試驗得出瑞德西韋10 d療程的不良反應多于5 d療程,且無顯著療效;世界衛生組織“團結實驗”初步結果顯示,瑞德西韋對降低重癥患者病死率或者減少住院時間影響很小或沒有影響[31]。雖然早期臨床前數據表明瑞德西韋可能對COVID-19具有潛在的活性,但對COVID-19到底有沒有療效還需要等待進一步的研究結果。
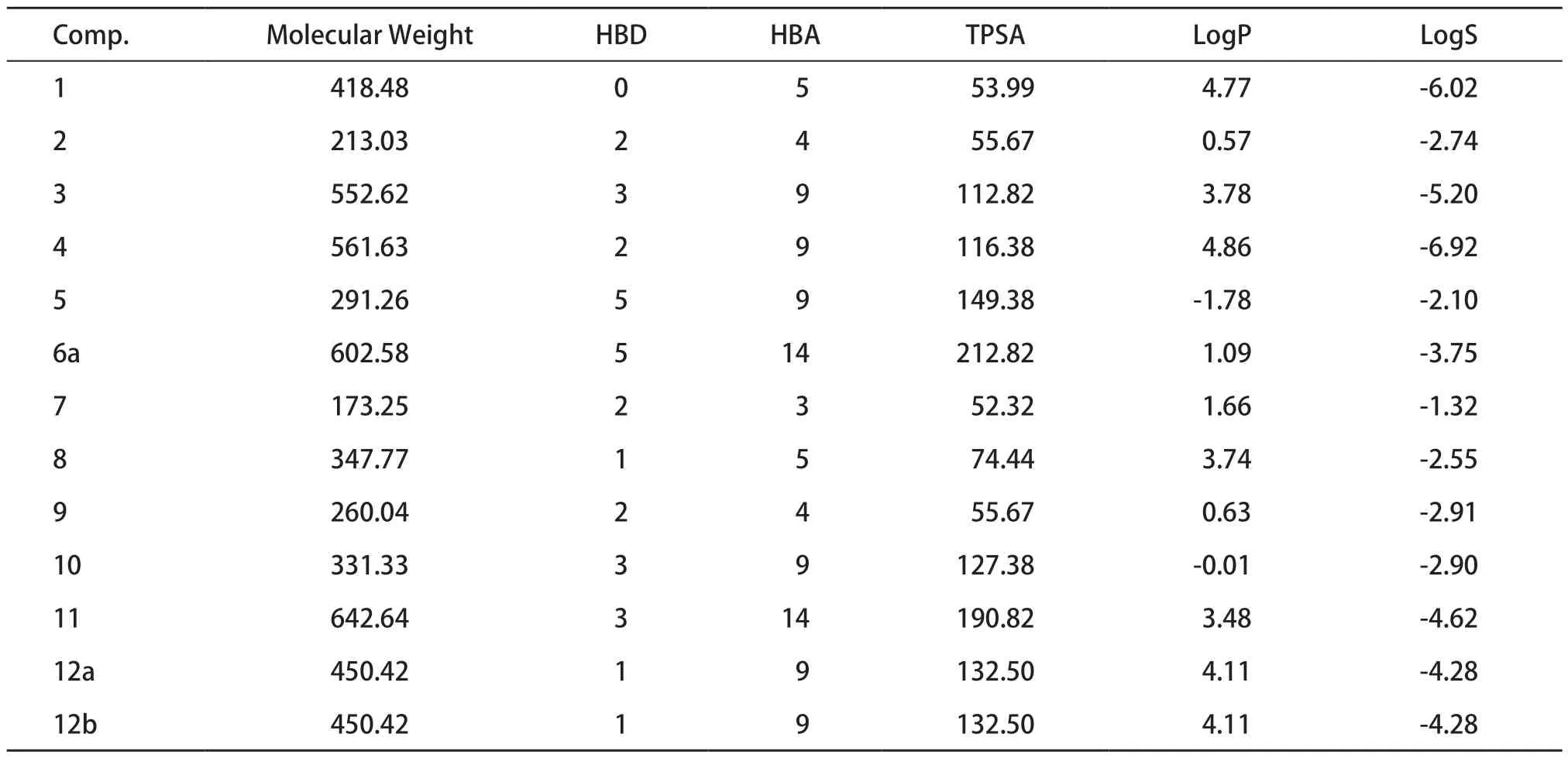
表3 瑞德西韋及其中間體13種化合物的理化性質
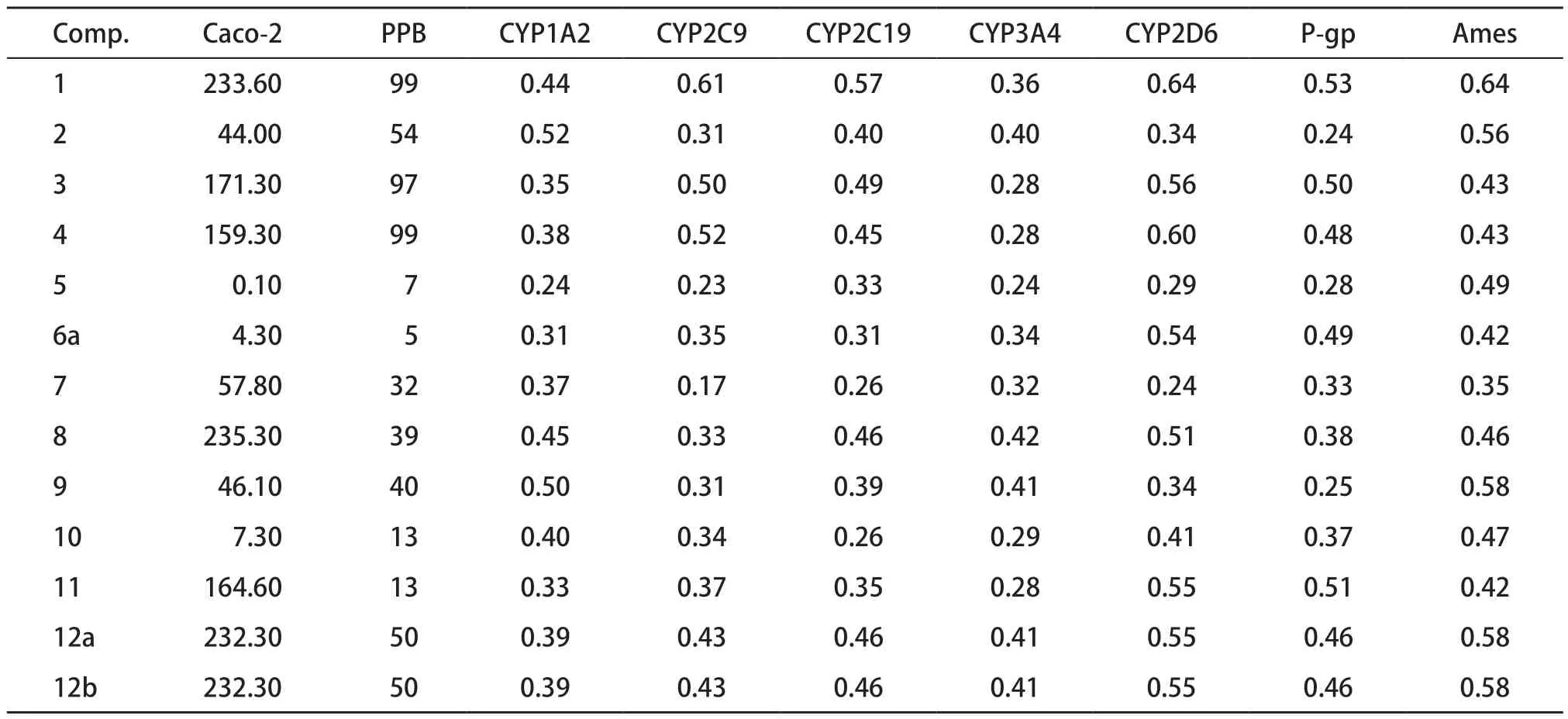
表4 瑞德西韋及其中間體13種化合物的藥代動力學參數
3 結論
本研究利用密度泛函理論對瑞德西韋及其合成中間體的分子結構、反應活性位點、紅外光譜以及概念密度泛函活性指數進行分析,并利用ACD/Percepta分析模型預測化合物的ADME/Tox。幾何結構分析表明計算方法是可靠的。分子表面靜電勢、原子電荷、前線軌道理論結果表明,戊糖環上的芐氧基或羥基氧易受到親電試劑的攻擊,鳥嘌呤類似物環上的氨基氫或支鏈胺基氫易受到親核試劑的攻擊,且鳥嘌呤類似物結構是瑞德西韋的母核結構,瑞德西韋及其具相同母核結構的化合物中,化合物5的活性較強。通過綜合對比化合物5和瑞德西韋(6a)的藥理活性,也表明了化合物5的活性優于瑞德西韋(6a)。最后通過ACD/Percepta分析預測13種化合物的理化性質和藥代動力學評價,推測瑞德西韋可能對COVID-19療效甚微或基本無效,以待進一步的大規模臨床試驗去評估其療效和安全性。本研究結果為揭示瑞德西韋及其合成中間體結構與性質的關系提供了理論基礎,也對深入探索瑞德西韋及其合成中間體生理活性,拓寬其在藥物研發中的應用提供了參考依據。
