伴肌動蛋白基因突變桿狀體肌病2型1例報告并文獻復習
2021-10-09 03:05:32黃坤玲劉建華牛波路素坤曹麗潔及立立呂文山帥金鳳
山東醫藥 2021年26期
關鍵詞:基因突變
黃坤玲,劉建華,牛波,路素坤,曹麗潔,及立立,呂文山,帥金鳳
河北省兒童醫院呼吸科,石家莊050031
先天性肌病是一組罕見的具有臨床、組織病理學和遺傳異質性的由肌纖維結構異常定義的遺傳性肌肉疾病。先天性肌病早期典型的臨床體征是肌張力減退、肌肉無力、營養不良和(或)運動障礙延遲,大多數情況下不影響中樞神經系統,智力正常[1-2]。桿狀體肌病(NM)是由編碼細肌絲相關蛋白的基因突變引起的顯性或隱性遺傳的先天性異質性肌病,其特征是骨骼肌無力,肌纖維中存在大量桿狀體。編碼骨骼肌α肌動蛋白(ACTA1)和伴肌動蛋白(NEB)基因突變是NM最常見的遺傳基因突變,其中由NEB基因突變引起的肌病被稱為NM2型[3]。國外對于NM的報道較多,國內多為個案報道,且大多經骨骼肌肌肉活檢病理確診,少數經基因檢測確診。2018年6月我院收治1例NM2型患兒,經NEB基因檢測確診。現總結其臨床特征及遺傳學特點,并復習相關文獻,以提高臨床醫生對該病的認識。
1 病例資料
患兒男,15歲,主因“肌無力、發育遲緩、反復呼吸道感染13年加重伴咯血、咳痰4 d”于2018年6月7日就診我院。患兒系第1胎第1產,足月順產,出生體質量3.5 kg,否認窒息缺氧史。患兒于2歲半時,家屬發現其走路姿勢呈鴨步態,智力語言發育大致正常,曾就診當地醫院,考慮肌營養不良,家屬拒絕進一步檢查;之后反復呼吸道感染,在當地醫院采取對癥抗感染治療。其弟13歲,臨床表現與哥哥相同(未予特殊治療),其他家族成員無類似疾病史,無家族遺傳病史,父母均體健,非近親結婚。
入院體格檢查:身高165 cm,體質量55 kg,體溫36.0℃,心率86次/分,呼吸26次/分,血壓103/74 mmHg,臉型瘦長,表情淡漠,脊柱后凸及輕度側彎,四肢肌力3級,肌張力低,雙側巴賓斯基征未引出,雙側膝腱反射、跟腱反射未引出,雙側肱二頭肌、肱三頭肌反射陽性。無震顫、手足徐動、舞蹈樣動作及感覺異常。
入院輔助檢查:血、尿、便常規正常,血清電解質、肝腎功能、心肌酶譜、血糖、血乳酸、血氨、血尿有機酸代謝篩查均正常,心電圖、心臟超聲、頭顱核磁未見異常。胸部高分辨CT檢查提示:右肺中葉、左肺上葉舌段可見多發囊狀含氣空腔,左肺下葉可見多發大小不等含氣空腔,并可見多個氣—液平面及雙軌征。
遺傳學檢查:經患兒家屬知情同意,于2018年6月16日采集患兒及其父母外周血標本各2 mL(家長因經濟原因未同意采集患兒弟弟外周血),送至北京邁基諾基因科技股份有限公司進行基因測序。提取基因組DNA建立全基因組文庫,采用液相捕獲技術對疾病相關基因的外顯子區域及其上下游50 bp區域進行捕獲,利用基因測序儀(美國Illumina公司,Hiseq2000型)進行高通量測序,平均深度不低于200×(二代測序在邁基諾醫學檢驗所完成)。根據靶向外顯子捕獲測序結果,對患兒及其父母DNA進行Sanger驗證,驗證引物為:上游5'-AATATC?CATTCATCAGAAATTGAGG-3',下 游5'-CATGCT?TATACCCTGCCTGTATTC-3'。通過二代測序,患兒NEB(NM_001271208)基因c.23122-1G>C雜合突變。根據美國醫學遺傳學與基因組學學會(ACMG)指南,PVS1(該突變為零效突變,可能導致基因功能喪失)及PM2(在正常人群數據庫中不出現,為低頻突變)兩條證據支持c.23122-1G>C位點判定為“可能致病”。同時發現NEB基因存在40~60號外顯子雜合缺失(見圖1),人類基因突變數據庫(HGMD)未見報道。根據ACMG指南,PVS1及PM2兩條證據支持40~60號外顯子雜合缺失突變判定為“可能致病”。通過Sanger測序對c.23122-1G>C位點進行家系驗證,驗證結果顯示,該突變來自于母親(見圖2)。40~60號外顯子雜合缺失的突變來源未做進一步驗證。
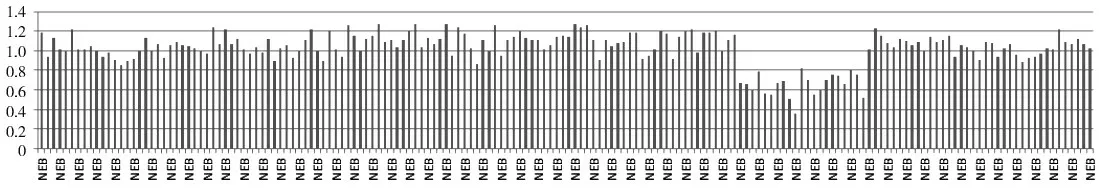
圖1 NEB基因40-60號外顯子雜合缺失圖
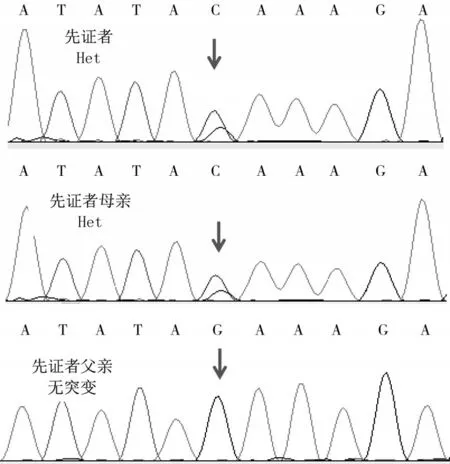
圖2 家系Sanger測序驗證圖
診療經過:入院后給予積極抗感染、祛痰及行支氣管鏡肺泡灌洗液等綜合治療,病情好轉出院。
2 討論
NM是一組罕見的先天性異質性肌病,編碼細肌絲相關蛋白的基因突變是其主要病因。文獻顯示,與NM相關的致病基因有13種,分別為ACTA1、NEB、TPM2、TPM3、TNNT1、KBTBD13、CFL-2、KLHL40、KLHL41、MYO18B、LMOD3、TNNT3、MYPN基因,其中后6種致病基因均為常染色體隱性遺傳[4]。ACTA1和NEB基因突變是NM最常見的致病基因。NEB基因位于2號染色體長臂區域,全長249 kb,含有183個外顯子,編碼人類肌節中最大的蛋白之一即NEB。NEB存在于肌節的I帶中,相對分子質量為700 kD,是細肌絲的組成部分,具有多個重復的肌動蛋白結合結構域,連接細肌絲與Z區,并將肌動蛋白單體連接在一起。NEB的作用是調節肌纖維成熟時肌動蛋白單體裝配成細肌絲的長度。由NEB基因突變引起的肌病為NM2型,其特征是早發性肌無力,軸向肌肉和近端肢體肌肉最為明顯,面部和延髓肌肉無力通常會導致構音障礙,患者的壽命取決于呼吸肌無力的嚴重程度[5]。對于某些患者,機械通氣是必要的,并且骨科并發癥很常見。NEB突變通常是復合雜合突變呈常染色體隱性遺傳,但是最近發現,一個家族的NM具有NEB的顯性遺傳突變[6]。本例患兒的NEB基因突變來源于母親的c.23122-1G>C雜合突變,內含子區第23122-1號核苷酸由鳥嘌呤突變為胞嘧啶,從而導致氨基酸發生改變,c.23122-1G>C雜合突變未見報道。同時分析到NEB基因存在40~60號外顯子雜合缺失。
2000 年歐洲神經肌肉病委員會國際協作組根據病情嚴重程度及年齡對NM進行臨床分型,主要分為以下6型[7]:①經典型:最常見,嬰兒期或兒童期起病,出生時有明顯的肌無力,四肢近端、面肌、呼吸肌、頸肌和咽喉肌也可累及,伴肌張力低下,運動發育遲緩,病情緩慢進展或靜止;②嚴重型:次常見,出生時即有嚴重的肌無力和(或)無自主呼吸,常累及心肌,病情嚴重且進展迅速,死亡率極高;③中間型:癥狀較嚴重型稍輕,病情呈逐漸加重趨勢,尚能自主活動,通過呼吸支持可維持生命;④輕型:癥狀輕,四肢無力少有或癥狀輕微,無雙足下垂和面部肌肉無力,兒童或青少年起病;⑤成人型:成人起病的為此類分型,少見,肌無力早期不伴肌肉形態學的改變;⑥其他類型:特征不典型,肌無力發生在不常見部位,眼肌麻痹等。NM的臨床表現異質性較大,新生兒和成年人均可發病,病情輕重不一,以對稱性的肌無力、腱反射減弱和肌張力減低為主,一般不影響中樞神經系統。疾病初期可僅有軀干肌和肢體肌肉無力,病情進展出現全身無力,多伴有肌肉萎縮,疾病后期常累及呼吸肌因呼吸衰竭而亡。本例患者為幼兒期起病,首先累及肢體近端,2歲半起走路姿勢呈鴨步態,于青少年期發展至全身無力,但病情相對穩定,屬于桿狀體疾病的經典型。
NM是累及多個系統的先天性疾病,許多系統性疾病與支氣管擴張有關,如系統性紅斑狼瘡、類風濕關節炎、強直性脊柱炎、干燥綜合征及復發性多軟骨炎等[8]。本患兒因累及呼吸肌出現反復呼吸道感染后致支氣管擴張就診呼吸科,經呼吸科確診。
病理診斷和基因診斷是NM診斷的金標準。病理診斷主要依靠肌肉病理活體組織檢查,在肌細胞胞質中找到桿狀體是該病特征性的肌肉病理改變。桿狀體通常出現在胞質尤其是肌膜下。隨著對NM認識的逐漸加深,發現桿狀體不一定就是NM,多發性肌炎及皮肌炎、線粒體腦肌病、強直性肌營養不良等疾病,也可發現桿狀體[9]。因此本病不能單獨依靠肌肉的病理檢查診斷。由于NM的遺傳、臨床異質性,不同的致病基因與疾病的表型和組織學表達有關,基因測序越來越多地用于NM的診斷。本例患兒肝酶及心肌酶譜正常,曾考慮肌營養不良,但家長拒絕進一步檢查,經反復勸說同意基因檢查,發現患兒NEB基因存在c.23122-1G>C雜合突變,同時NEB基因的40-60號外顯子存在雜合缺失,依靠臨床表現和基因檢查,確診為NM2型。該病需與皮肌炎、多發性皮肌炎、線粒體腦肌病、強直性肌營養不良等鑒別[10]。皮肌炎、多發性皮肌炎患者肌酶明顯升高,肌肉組織活檢可見大量炎癥細胞浸潤,皮肌炎常可累及皮膚,伴有明顯的皮損;而NM患者肌酶正常,肌肉組織中炎癥細胞浸潤較少。線粒體腦疾病是一組線粒體相關的代謝性多系統疾病,主要累及腦、骨骼肌及心肌組織,臨床主要表現為頭痛、反復癲癇發作、肌無力,運動后特征性乳酸升高[11];本例患兒無此特征。強直性肌營養不良多為成人期起病的常染色體顯性遺傳疾病,主要表現為肌無力、肌強直和肌萎縮[12];本例患兒于幼兒期起病。
綜上所述,對于幼兒期起病、病程較長,表現為肌無力、肌張力低下累及多系統疾病的患者,需考慮NM可能,進一步行肌肉活體組織病理檢查和基因檢測以明確診斷,從而協助指導優生優育。NM診斷的金標準是病理檢查和基因檢測。本患兒NEB基因c.23122-1G>C雜合突變為新發現的突變位點。
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中國醫學影像學雜志(2021年6期)2021-08-13 08:43:36
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學生導刊(2018年13期)2018-06-29 03:49:00
中國生殖健康(2018年2期)2018-01-12 13:57:51
現代檢驗醫學雜志(2016年4期)2016-11-15 02:01:14
中國現代醫學雜志(2015年26期)2015-12-23 11:04:22
鄭州大學學報(醫學版)(2015年2期)2015-02-27 14:50:44
中華皮膚科雜志(2014年4期)2014-12-19 12:55:49
中國神經精神疾病雜志(2014年1期)2014-03-01 03:23:22
