血清淀粉樣蛋白A通過LOX-1促進系膜細胞攝取脂質
2021-08-19 03:22:36郝怡然陳逸歌劉夢妤
醫學研究雜志 2021年8期
郝怡然 陳逸歌 李 彧 劉夢妤 劉 華
臨床研究發現各種腎臟疾病常伴有炎性反應,而炎癥促進腎小球硬化的發生、發展[1]。血清淀粉樣蛋白A(serum amyloid A,SAA)作為強效炎性因子,在炎癥和脂質代謝中起關鍵作用[2]。SAA檢測有助于多種疾病的診斷和預后評估,如類風濕關節炎、動脈粥樣硬化、肥胖、糖尿病腎病(diabetic kidney disease,DKD)[3~6]。最近的證據顯示SAA是炎癥通路的中樞介質,SAA水平在DKD早期升高,促進腎小球硬化和腎間質纖維化[7]。高脂血癥常伴發于多種腎臟疾病,如腎病綜合征,是促進腎小球硬化的危險因素[8]。炎性因子可能與脂質失衡共同參與了腎小球硬化的進展,觀察炎性因子對細胞脂質穩態的影響是探討脂質介導腎損傷的關鍵[9]。氧化低密度脂蛋白(oxidised low-density lipoprotein, Ox-LDL)植物血凝素樣受體1(lectin-like oxidised low-density lipoprotein receptor 1, LOX-1)在炎癥和脂質代謝紊亂相關性疾病的發生、進展中發揮重要作用[10~12]。炎性因子SAA對人腎小球系膜細胞(human mesangial cells,HMCs)LOX-1受體及脂質代謝的相關研究較少。本研究通過觀察SAA對HMCs攝取脂質及LOX-1受體表達的變化,探討炎癥在細胞水平影響HMCs脂質穩態的病理、生理機制。
材料與方法
1.材料:RPMI1640培養液及胎牛血清(Foetal calf serum,FCS)(美國Gibco Life Technologies 公司),人腎小球系膜細胞由英國皇家醫學院腎臟病中心阮雄中教授惠贈,SAA(美國Peprotech Technologies公司)、Ox-LDL(中國醫學科學院基礎研究所)、油紅“O”(美國Sigma-Aldrich公司),熒光Dil-Ox-LDL (美國Biomed Technologies公司)、LOX-1抗體(美國R&D Systems公司)。PCR反應體系、反轉錄酶緩沖液(美國Promega Corporation公司)、dNTP、Oligo(dT)、引物(上海生物工程有限公司)合成,LOX-1引物序列:上游引物:5′-ACAGAGGCCATTCCGAAATCA-3′,下游引物:5′-GGTAGAGTCTGGAGATGGACCACA-3′。GAPDH 引物序列:上游引物:5′-GCACCGTCAAGGCTGAGAAC -3′,下游引物:5′-ATGGTGGTGAAGACGCCAGT -3′。
2.HMCs的培養:用10%FCS的RPMI1640培養液在5%CO2培養箱37℃培養HMCs,0.01mol/L PBS沖洗,0.25%胰蛋白酶/0.025 EDTA消化,10%FCS的RPMI1640培養液懸浮細胞傳代培養。
3.油紅“O”染色:培養HMCs (5×104個/毫升)至亞融合狀態,靜止后將細胞隨機分組,對照組(0.2% RPMI1640)和SAA處理組(5ng/ml SAA+0.2% RPMI1640),加入Ox-LDL(40mg/L)繼續培養24h,油紅“O”染色后觀察攝片。
4.流式細胞計數:培養HMCs至亞融合狀態,靜止后將細胞隨機分組,對照組、SAA組(5ng/ml SAA)和LOX-1受體阻斷+SAA組(2ng/ml 抗LOX-1抗體+5ng/ml SAA,抗LOX-1抗體提前2h應用),孵育12h各組加入10mg/L的熒光Dil-Ox-LDL,繼續培養5h。PBS重復沖洗、消化、離心,制成細胞懸液,流式細胞儀計數6000個細胞,計算各組細胞內平均熒光值。
5.實時定量PCR測定:培養HMCs至亞融合狀態,靜止后隨機分為3組,分別用含0、5、10ng/ml SAA的培養基,培養12h后收獲細胞,提取總RNA,合成cDNA,25μl反應體系進行PCR擴增,95℃10s,95℃15s,60℃30s,共擴增45~50個循環。
6.Western blot法檢測:培養HMCs至亞融合狀態,靜止后隨機分為3組,分別用含0、5、10ng/ml SAA的培養基,培養12h后收獲細胞,提取總蛋白。上樣100μg蛋白質電泳,轉膜1h,封閉緩沖液過夜,0.2ng/L一抗與膜孵育,室溫緩慢振蕩2h,洗膜,1∶5000二抗室溫緩慢振蕩1h,洗膜,ECL顯影。
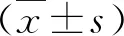
結 果
1.SAA促進HMCs對Ox-LDL的攝取:油紅“O”染色發現HMCs攝取Ox-LDL,SAA組較對照組細胞內紅染顆粒明顯粗大、增多,提示SAA增強HMCs對Ox-LDL的攝取(圖1)。
圖1 SAA(5ng/ml)促進HMCs對Ox-LDL的攝取(油紅“O”染色,×200)A.對照組;B.SAA刺激組
2.流式細胞儀計數:HMCs攝取Dil-Ox-LDL,對照組HMCs平均熒光值為8.6,SAA組細胞內平均熒光值為51.9,為對照組的6.03倍,提示SAA促進HMCs攝取Dil-Ox-LDL,應用抗LOX-1共孵育2h,阻斷組細胞內平均熒光值為41.2,為對照組的4.79倍,降低為單純SAA刺激組的79.3%(圖2)。
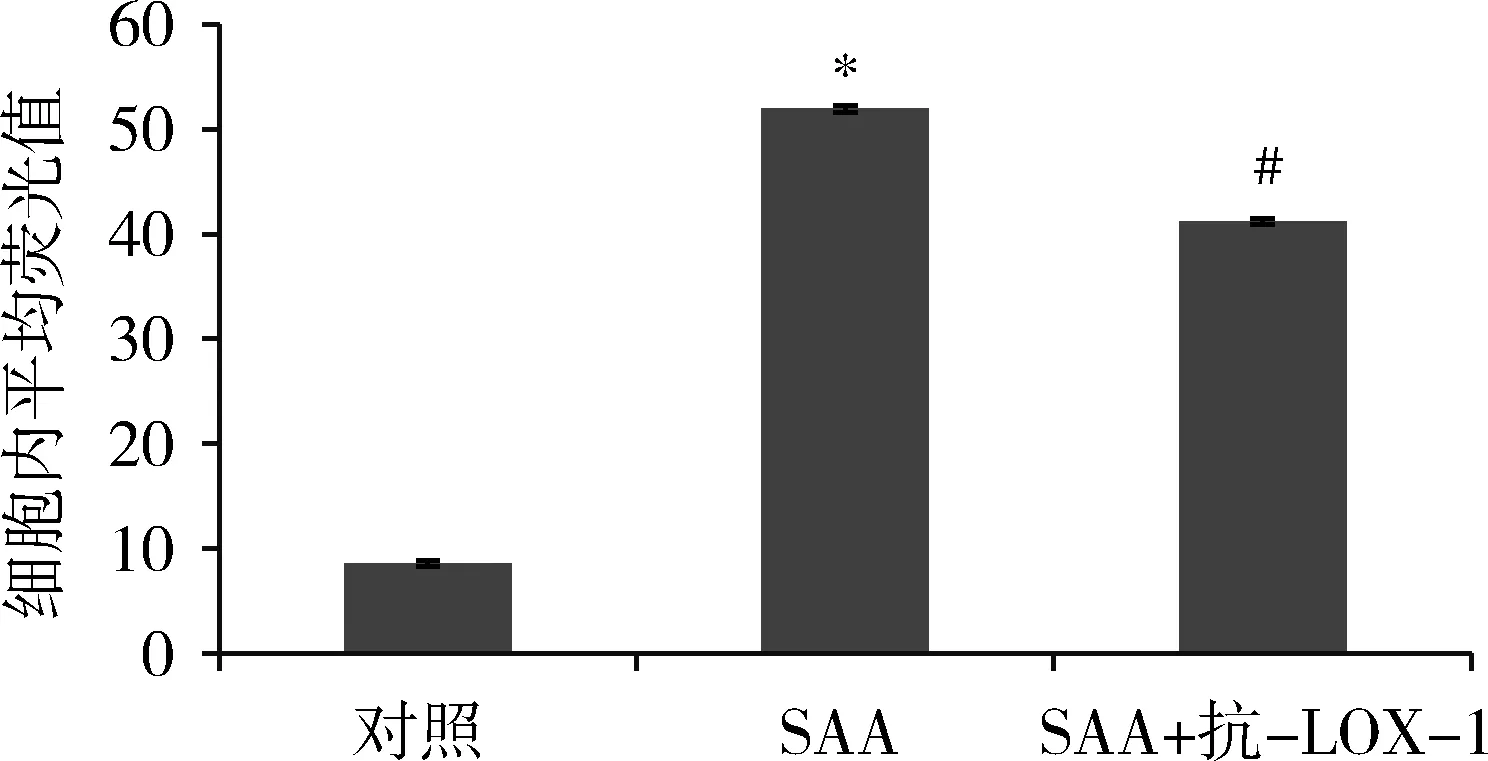
圖2 SAA刺激及抗LOX-1抗體對HMCs攝入Dil-Ox-LDL的影響與對照組比較,*P<0.05;與SAA組比較,#P<0.05
3.SAA對LOX-1 mRNA表達的影響:SAA促進LOX-1 mRNA的表達。用0、5、10ng/ml SAA刺激HMCs 12h,5ng/ml SAA組LOX-1 mRNA水平升高為對照組的5.12倍,10ng/ml SAA組LOX-1 mRNA水平達高峰,為對照組的6.97倍(圖3)。
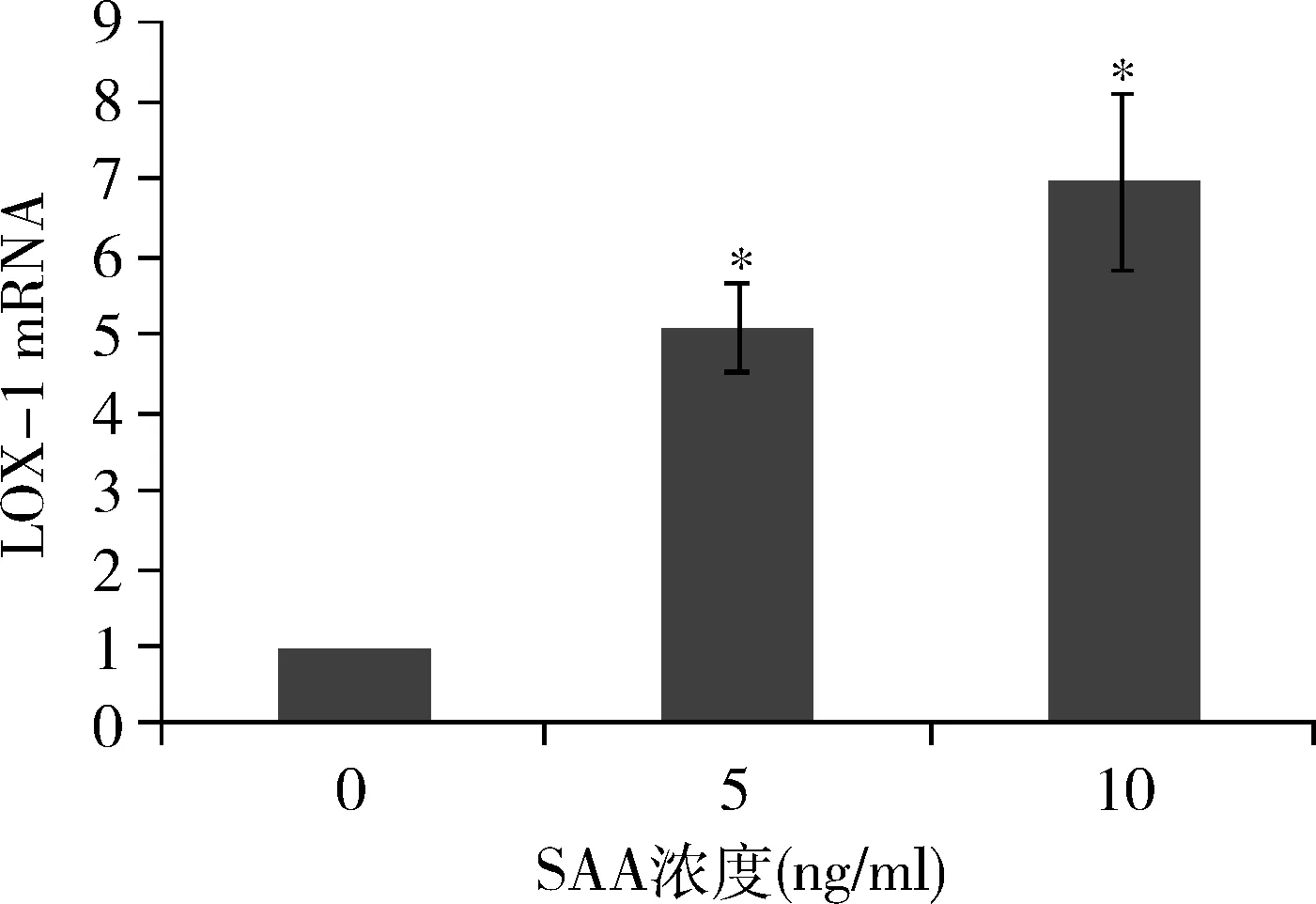
圖3 不同濃度SAA作用12h對LOX-1 mRNA表達的影響與0ng/ml比較,*P<0.05
4.SAA對LOX-1 蛋白表達的影響:SAA促進LOX-1蛋白的表達。用0、5和10ng/ml SAA刺激HMCs 12h,5ng/ml SAA組LOX-1蛋白水平升高為對照組的2.61倍,10ng/ml SAA組LOX-1蛋白水平達高峰,為對照組的3.15倍(圖4)。
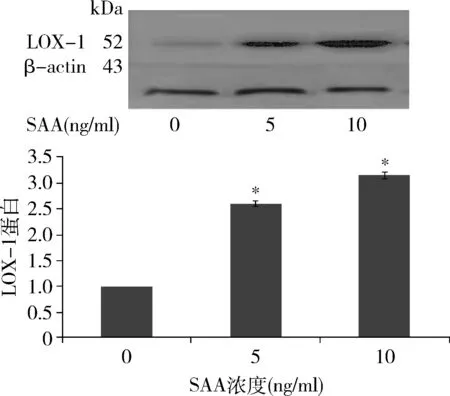
圖4 不同濃度SAA作用12h對LOX-1蛋白表達的影響與0ng/ml比較 *P<0.05
討 論
研究表明,原發和繼發性腎臟疾病常伴發炎性狀態,如IgA腎病、狼瘡性腎炎,抗炎治療是有效的治療措施[13]。SAA作為急性時相反應蛋白,在炎性反應中迅速升高,并參與慢性炎性疾病的發展。流行病學研究發現,類風濕關節炎、代謝綜合征、冠狀動脈粥樣硬化和DKD患者中SAA水平顯著升高[14~17]。近年來研究發現SAA誘導巨噬細胞形成泡沫細胞,抑制膽固醇的逆向運輸,導致細胞內脂質失衡,促進動脈粥樣硬化的形成[18]。因此炎性因子SAA與高脂血癥共同參與了動脈粥樣硬化的發病機制。腎臟疾病常表現為炎性狀態,高脂血癥是腎臟疾病的獨立致病因素,因此研究炎癥導致細胞脂質失衡的機制是闡明脂質介導腎損傷的關鍵路徑,目前炎性因子SAA是否影響HMCs脂質穩態尚不明確。
SAA可能通過LOX-1途徑促進HMCs攝取Ox-LDL影響細胞內脂質穩態。既往研究表明腎小球硬化與動脈粥樣硬化類似,是多種細胞因子參與的慢性炎癥過程。HMCs在生理條件下攝取Ox-LDL,通過自我調節與脂質代謝相關受體的表達,維持細胞內脂質平衡。炎性因子可以打破脂質水平介導的負反饋調節,導致大量攝入脂質,最終超過其清除能力形成泡沫細胞[19]。高脂血癥的程度與腎小球硬化的程度不平行,近期研究顯示炎癥是加重脂質異常導致腎損害的關鍵[20]。本研究表明,炎性因子SAA促進HMCs攝取大量Ox-LDL,脂質過度積聚導致泡沫細胞形成,HMCs來源的泡沫細胞可進一步釋放細胞因子導致腎小球硬化,提示炎性因子SAA在細胞水平影響脂質穩態[21]。LOX-1作為Ox-LDL的重要受體,能特異性結合Ox-LDL,在內皮細胞、巨噬細胞、血管平滑肌細胞、成纖維細胞和其他細胞中廣泛表達。既往研究已經證實,Ox-LDL可以正反饋刺激自身LOX-1受體,炎性因子進一步促進LOX-1表達,LOX-1-Ox-LDL通路可激活核因子-κB(NF-κB)等細胞因子,引發瀑布級聯樣炎性反應[22]。本研究表明,SAA促進HMCs攝取Ox-LDL,而抗LOX-1抗體可部分阻斷對Ox-LDL攝取,提示SAA可通過LOX-1受體途徑刺激HMCs攝取大量Ox-LDL。因此,SAA促進HMCs通過LOX-1途徑攝取過度脂質,誘導轉化生長因子-β等細胞因子表達,促進腎小球細胞增殖,提示LOX-1在腎小球硬化進展中發揮關鍵作用[23]。
LOX-1是Ox-LDL的重要受體和關鍵炎性因子,SAA作為強有力的炎性介質促進LOX-1表達。研究發現SAA作為炎性因子在多種組織和細胞發揮促炎作用,導致代謝紊亂和動脈粥樣硬化的形成。通過對肥胖患者的觀察發現,SAA水平可間接反映胰島素抵抗的程度。SAA可以抑制膽固醇的逆向運輸升高血漿膽固醇濃度,進一步促進動脈粥樣硬化的形成。因此,SAA作為代謝綜合征和炎癥相關疾病的危險因素得到廣泛重視。越來越多的證據表明,炎癥是DKD發生、發展的關鍵環節,SAA與DKD患者組織學嚴重程度相關,顯著參與DKD的進展。SAA在高糖條件培養的足細胞和系膜細胞表達上調,進一步誘導炎性反應促進DKD特征性損害。因此,SAA介導的炎癥信號通路成為DKD新的生物學標志物和潛在的治療靶點。SAA結合高密度脂蛋白通過NF-κB途徑刺激低密度脂蛋白受體缺陷小鼠腎臟LOX-1表達,促進腎臟纖維化。高糖通過磷酸化絲裂原活化蛋白酶P38途徑誘導LOX-1的表達,增強Ox-LDL誘導的腎小管間質細胞凋亡,為DKD的分子機制研究提供了新思路。抗LOX-1治療可能是逆轉血脂異常和DKD致病因素的有效方法,因此LOX-1在糖尿病和慢性腎臟病進展中發揮關鍵作用[23]。本研究提示SAA可能通過LOX-1途徑增強Ox-LDL攝取,促進LOX-1表達,導致HMCs脂質失衡轉化為泡沫細胞,表明與動脈粥樣硬化發病機制類似,SAA水平升高是腎小球硬化的危險因素,阻斷或拮抗SAA和LOX-1相關炎癥通路可能延緩DKD進展。
綜上所述,炎性因子SAA通過LOX-1受體途徑促進Ox-LDL攝取,而抗LOX-1抗體可降低對Ox-LDL攝取,SAA進一步促進LOX-1 mRNA和蛋白的表達。因此,炎性因子SAA通過增強LOX-1受體表達促進脂質的攝取,影響細胞內脂質穩態,進一步可激活多種細胞因子,促進炎性反應加重脂質腎損害。SAA水平升高可能是DKD發展的危險因素和生物學標志物,然而仍需進一步研究闡明具體致病機制,阻斷相關炎癥通路及脂質代謝紊亂將是治療腎小球硬化的新靶點。
