鹽酸法磷酸萃取相的洗滌和磷酸反萃取過程初探
2021-08-12 07:21:10張少杰唐盛偉鄭東鑰鐘本和陳彥逍
無機鹽工業 2021年8期
關鍵詞:實驗
張少杰,唐盛偉,王 楊,鄭東鑰,鐘本和,陳彥逍
(四川大學化學工程學院,四川成都610065)
磷酸是磷化學工業中的重要化工產品,被廣泛應用于農業、食品、制藥、電子工業等國民經濟部門。根據其制備過程不同,磷酸主要分為熱法磷酸和濕法磷酸。其中熱法磷酸根據其純度可分為工業級磷酸、食品級磷酸、試劑級磷酸以及電子級磷酸,而濕法磷酸雜質含量較高,據統計90%以上的濕法磷酸都用于磷肥生產[1]。
濕法磷酸過程主要是指利用無機強酸(硫酸、鹽酸、硝酸)分解磷礦制備磷酸,相應的濕法磷酸路線又以硫酸分解磷礦結合溶劑萃取凈化磷酸較為成熟,國外在20世紀60年代后對溶劑萃取凈化濕法磷酸的技術進行開發,其中以色列礦業公司(IMI)、德國巴登哈姆公司(Budenheim)、比利時普雷昂公司(Prayon)、英國奧布拉威爾森公司(Albright&Wilson)以及法國羅納-普朗克公司(Rhone-Poulenc)等形成了專利和工業化流程[2];而中國于2003年由四川大學和貴州宏福實業有限公司開發了適合中國濕法磷酸品質的凈化技術,并獲得了合格的凈化濕法磷酸產品[3]。
目前,國內主要使用硫酸法過程生產濕法磷酸:
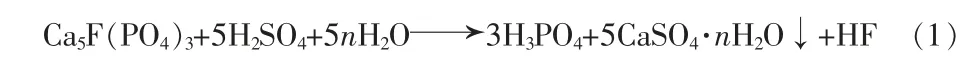
硫酸法過程中消耗硫酸的費用約占直接成本的60%[4];同時每生產1 t磷酸(P2O5計)副產4.5~5.0 t磷石膏,目前世界年磷石膏排放量為1.6億~1.7億t[5]。中國年排放量在2017年末已達7 500萬t,對磷石膏綜合利用率的要求在“十二五”之后已經提高到30%以上,但由于磷石膏中有害雜質含量高、開發利用投資較大、后沿產品附加值低、利潤微薄,導致了不少企業磷石膏利用裝置開工率低,存在“以銷定產”的情況,大量磷石膏仍以堆存為主,占用土地資源的同時更導致環保安全出現隱患[6-7]。
中國磷礦質量較差,中低品位礦多,富礦少,全國磷礦平均品位在17%(P2O5)左右,其中富礦占磷礦總量約為8.5%[8]。而根據HG/T 2673—1995《酸法加工磷礦石》和HG/T 2674—1995《黃磷用磷礦石》,相應的濕法和熱法磷酸用磷礦品位均需達到24%(P2O5)以上。綜上,開發出低能耗、低成本、環境友好并且適用于中國中低品位磷礦的磷酸生產工藝尤為關鍵。
窯法磷酸工藝和鹽酸法濕法磷酸工藝在中低品位磷礦利用方面具有其獨到的優勢。基于與熱法磷酸相同的工藝原理,中國目前研發的窯法磷酸工藝可以利用中低品位硅質磷礦制備出純度與熱法酸相當的磷酸,且具有更好的經濟效益,但由于技術原因中國的窯法磷酸仍停留在工業試生產階段[9-10]。
而鹽酸法濕法磷酸的優勢在于可利用工業副產鹽酸(質量分數為20%~35%)對高硅中低品位磷礦進行分解,分解速率快,磷收率高,其主化學反應式:

針對鹽酸法工藝,以色列礦業公司(IMI)開發了相應的專利技術,實現了磷酸與CaCl2的分離,并推行工業化,用于生產飼料級和食品級磷酸。但由于IMI流程使用的混合醇類萃取劑(體積分數為60%正戊醇+40%異戊醇),溶劑回收的工序更增加了流程的復雜程度與能耗[11]。
磷酸三丁酯(TBP)被認為是一種優良的磷酸萃取劑,其具有性質穩定、水中溶解度小、對磷酸萃取效力強、對雜質凈化程度高等優點。四川大學基于TBP開發出了免溶劑回收的濕法磷酸萃取凈化工藝,每1 t P2O5溶劑消耗量僅為6 kg,具有良好的經濟效益和工藝可行性[3]。李軍等則對TBP在鹽酸法體系中萃取凈化濕法磷酸進行了研究,結果表明磷酸在TBP相中的分配系數隨著水相中CaCl2的濃度提高而提高,TBP在鹽酸法流程中具備良好的應用前景[12-13],并基于此提出了以TBP作為主要磷酸萃取劑,通過鹽酸分解中低品位磷礦制備工業級和食品級磷酸的方法[14]。
鹽酸法磷酸工藝的關鍵在于通過液-液萃取過程分離磷礦浸出液中的H3PO4和CaCl2,由于萃取H3PO4時少量CaCl2被同時萃取,需要綜合考慮過程中H3PO4萃取、有機相洗滌、H3PO4反萃取3個環節,才可能最大限度地降低反萃酸中的CaCl2含量,再經過后處理使凈化磷酸達到符合GB/T 2019—2008《工業磷酸》規定的氯含量標準。當以TBP萃取凈化鹽酸法工藝得到粗磷酸時,由于萃取有機相的黏度較大,以小相比的磷酸或水相逆流洗滌有機相去除CaCl2的操作存在困難,故采取了以一定濃度的H2SO4化學洗滌有機相去除CaCl2的方法。目前,國內外關于溶劑萃取凈化濕法磷酸的研究多集中于萃取劑選取及萃取過程等方面,而針對有機相的洗滌與反萃取過程少見詳細報道。筆者主要針對以TBP萃取鹽酸法磷酸所得有機相的洗滌和磷酸反萃取過程進行初步探究,重點討論不同洗滌、反萃方法和條件對反萃酸品質以及硫酸鈣的結晶過程的影響,從而為開發以TBP為主要萃取劑的鹽酸法磷酸凈化工藝提供一定的指導。
1 實驗部分
1.1 主要實驗試劑
表1 為主要實驗藥品和試劑。

表1 主要實驗藥品和試劑Table 1 Main experimental drugs and reagents
1.2 實驗所用磷礦及其相關分析
實驗所用磷礦產自四川會東某地,其組成按照GB/T 1868、1870~1881—1995《磷礦石與磷精礦化學分析方法》系列國家標準進行測定[15],結果見表2;實驗所用磷礦-鹽酸浸出液為上述磷礦與質量分數為20%HCl按照理論量反應制得,其中H3PO4與CaCl2分別按照上述國標通過磷鉬酸喹啉重量法以及EDTA容量法測定,其余元素含量由離子發射光譜法(ICP-OES)進行測定,其組成見表3;實驗所用待反萃有機相為萃取劑(TBP與磺化煤油按照體積比4∶1均相混合)與磷礦浸出液(除鐵)達到萃取平衡所得,其組成見表4。

表2 實驗用磷礦組成(干基)Table 2 Composition of the experimental phosphate rock powder(dry basis) %
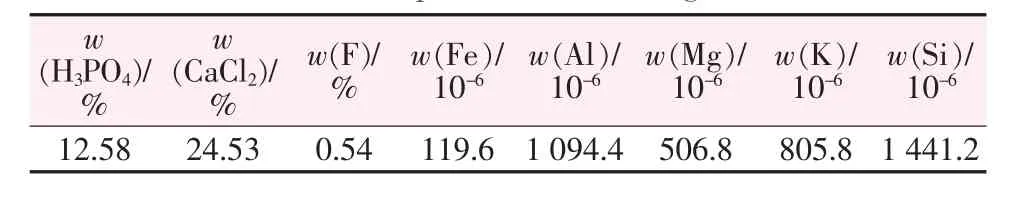
表3 鹽酸法磷礦浸出液組成(萃取除鐵后)Table 3 Composition of the phosphate ore-hydrochloric acid leach liquor(after removing Fe3+)

表4 待洗滌-反萃有機相的組成Table 4 Composition of the organic phase to be scrubbed and stripped %
1.3 主要試驗儀器
DX-2700 X射線衍射儀;JSM-7500F掃描電子顯微鏡—能譜儀;ICP-OES 5100 SVDV電感耦合等離子發射光譜儀;Particle Track G400聚焦光束反射測量儀;FZ10蠕動泵。
2 實驗結果與討論
2.1 一步法反萃取H3PO4實驗
通常,從磷礦-鹽酸浸出液中萃取H3PO4時,較高的CaCl2濃度會增大H3PO4在有機相中的分配系數,促使H3PO4被萃取進入有機相,而在用純水反萃有機相中的H3PO4時,CaCl2的濃度降低,又有利于H3PO4從有機相中萃出進入水相,提高H3PO4的回收率。本部分實驗使用一定體積的硫酸溶液同時作為CaCl2沉淀劑和H3PO4反萃取劑,在反萃H3PO4的過程中同時與CaCl2發生反應,過程中產生的HCl可以在后續鹽酸解析以及磷酸濃縮的過程中基本除凈:

實驗過程:取一定體積和質量的待反萃有機相,根據CaCl2和H2SO4的理論反應量,按照一定反萃取相比V(A)/V(O)(反萃取水相與待反萃有機相的體積比)配制相應濃度的稀硫酸,一次混合后在攪拌釜中進行恒溫反萃取。在CaSO4-H3PO4-H2O三元系統中,由于半水硫酸鈣(CaSO4·0.5H2O)形成晶核所需活化能最小,所以硫酸鈣首先是以非穩態的半水物生成的,而在較低的磷酸濃度下,半水物將轉化為二水物(CaSO4·2H2O)和無水物(CaSO4)兩種穩 定晶型[8,16]。考 慮 到式(3)的 反 應 程 度 和 硫 酸 鈣結晶的轉化動力學,反萃實驗在400 r/min下進行70 min,靜置后分去上層有機相,再通過離心和抽濾分離反萃酸中的石膏固相。石膏在45℃下干燥36 h后用作SEM和XRD分析。反萃酸中的H3PO4含量以磷鉬酸喹啉重量法測定,少量Ca2+通過離子發射光譜方法(ICP-OES)測定,最終H3PO4的反萃取率(SE)按照下式計算:

式中,maq、m0,org為反萃后水相和待反萃有機相的質量,g;w(H3PO4)aq、w(H3PO4)0,org為反萃后水相和待反萃有機相中H3PO4的質量分數,實驗結果見表5。由表5可知,當使用理論量的H2SO4對有機相進行反萃時,H3PO4的反萃取率隨著反萃取劑用量增加而升高,等體積[V(A)/V(O)=1∶1]反萃取時,H3PO4的反萃取率超過90%;溫度升高有利于H3PO4的反萃取。相較于待反萃有機相,發現反萃水相中的磷鈣比[n(H3PO4)/n(Ca2+)]顯著提升,說明了硫酸作為反萃取劑對Ca2+有比較明顯的去除效果,但由于反萃取磷酸中硫酸鈣溶解平衡的存在,水相中的Ca2+含量始終略高于0.2%(CaCl2質量分數)。且隨著反萃相比V(A)/V(O)的增大,反萃酸中H3PO4的反萃取率升高但是磷鈣比n(H3PO4)/n(Ca2+)下降,這同樣是硫酸鈣溶解程度增大所致。

表5 不同條件下以H2SO4溶液直接反萃H3PO4的實驗結果Table 5 Experimental results of H3PO4 stripping when H2SO4 solution was used as stripping agent under different conditions
圖1 為不同實驗組所得石膏的SEM照片。由圖1可見,在理論H2SO4用量下,當體系中初始H2SO4濃度降低時,所得石膏的結晶尺寸變大,形貌逐漸向板狀和長條狀過度,但在攪拌作用下出現細小碎晶,所得石膏的規整度變差,該現象不利于實際生產中石膏過濾的進行。其原因是SO42-濃度下降導致了水相中硫酸鈣結晶過飽和度降低,限制了結晶過程初期的成核數量,從而使晶體的幾何尺寸趨向于增大,在攪拌下機械碰撞效應也更為明顯,導致了晶體二次成核的發生和細晶的產生[17]。因此,通過控制硫酸濃度來控制結晶過飽和度,通過控制攪拌強度來減少結晶過程中的二次成核,對得到粗大均勻、易于過濾分離的硫酸鈣結晶尤為重要。

圖1 反萃取過程中生成CaSO4·nH2O的SEM照片Fig.1 SEM images of the prepared CaSO4·nH2O during extraction process
圖2 為實驗所得石膏的XRD譜圖。由圖2可以看出,所得石膏一般為無水、半水和二水的混合物。由CaSO4-H3PO4-H2O體系平衡圖可知,在較高溫度時(>40℃),硫酸鈣晶型的轉化順序為半水→二水→無水,溫度較低時(<40℃),轉化順序為半水→無水→二水[16]。實驗結果表明,隨著反應溫度的提高,無水硫酸鈣的衍射峰更加明顯。同時,隨著反萃硫酸初始濃度的減小,相同溫度和時間條件下,半水硫酸鈣的轉化程度明顯提高,在實驗組a3條件下,半水硫酸鈣已經基本完全轉化。

圖2 反萃取過程中生成CaSO4·nH2O的XRD譜圖Fig.2 XRD pattern of the prepared CaSO4·nH2O during extractin process
通過上述實驗結果可知:1)在用理論量的H2SO4對有機相中的H3PO4進行反萃時,提升反萃相比V(A)/V(O)、升高反萃取溫度可以提升H3PO4的反萃取率;2)降低反萃H2SO4的初始濃度可以得到幾何尺寸更大的硫酸鈣結晶,但此時晶體的破碎和二次成核現象也更為明顯,導致大量碎晶的產生,因此需要對反萃硫酸濃度和攪拌強度加以控制來得到粗大均勻、易于過濾的硫酸鈣結晶;3)由于受到硫酸鈣溶解化學平衡的限制,通過H2SO4溶液直接反萃有機相中的H3PO4無法進一步提高H3PO4和CaCl2的分離程度,因此下文中提出了CaCl2洗滌-H3PO4反萃的二步法流程,即先用少量H2SO4溶液洗滌有機相,分離掉硫酸鈣石膏后,再以軟水反萃取有機相中H3PO4,從而進一步降低Ca2+在反萃磷酸中的含量。
2.2 洗滌-反萃兩步法實驗
2.2.1 洗滌實驗
在使用理論量H2SO4對有機相進行洗滌去除CaCl2的過程中,為了降低被洗水帶走的H3PO4損失,采用了小相比[V(A)/V(O)=1∶10]進行洗滌。該過程較為復雜,涉及到硫酸液滴在有機相中的分散、H2SO4與CaCl2在液滴和有機相界面間的傳質和反應、CaSO4·nH2O晶體的非均相成核以及生長等過程,因而研究難度較大。
聚焦光反射測量技術(FBRM)可以用來研究分散在液體中的液滴或者微粒,獲取其尺度和數量的信息。圖3[18]為FBRM的工作原理,在研究液體中的結晶過程時,聚焦光束運動方向上掃過晶體微粒的距離被記錄為微粒的弦長(chord length),不同弦長區間內微粒的計數每個檢測掃描周期內檢測到微粒的個數為弦長分布(CLD),弦長分布可以一定程度上反映應晶體生成過程中的晶體尺度分布(CSD)變化。實驗中采用聚焦光束反射測量儀對洗滌過程進行研究,對有機相中硫酸鈣晶體的生成規律進行在線觀測。實驗原理見圖4,實驗溫度為20℃,攪拌轉速為400 r/min,調節蠕動泵轉速使洗滌硫酸在1 min時完成進料,進料開始同時以聚焦光束反射測量儀記錄結晶過程數據。

圖3 聚焦光反射測量儀檢測結晶過程原理圖[18]Fig.3 Crystallizing process observed by FBRM[18]

圖4 洗滌過程實驗原理圖Fig.4 Experimental schematic diagram of the scrubbing process
由于FBRM方法可以同時檢測到分散在有機相中的水相微液滴和洗滌過程中生成的硫酸鈣結晶。故洗滌試驗前先進行了空白實驗以確定液滴分散過程對結晶檢測的影響,即在相同的實驗條件下測試了硫酸在未萃取H3PO4和CaCl2的空白有機相(萃取劑組成80%TBP+20%煤油,體積分數)中的分散情況,相比為V(A)/V(O)=1∶10,結果見圖5。圖5表明,洗滌酸在空白有機相中主要形成了弦長<10 μm和10~50 μm的微液滴,二者計數和變化趨勢基本相當,均隨攪拌時間的增加而增加,30 min時趨于平穩;空白實驗中基本未見大液滴的生成,說明了在攪拌轉速和加料相比下洗滌硫酸在有機相中的分散良好。

圖5 反萃硫酸水溶液在有機相中的分散實驗(空白實驗)Fig.5 Dispersion experiment of sulfuric acid droplets in organic phase(blank experiment)
圖6 為洗滌過程中硫酸鈣晶體生成的規律。由圖6可知,在開始勻速進料洗滌硫酸時,有機相中立刻出現大量弦長<10 μm和10~50 μm的微粒,其計數遠高于圖5中洗滌硫酸在空白實驗中有機相中分散成微液滴時的計數,說明此時有機相中生成的結晶微粒數量占主導地位,液滴的分散對結晶過程的檢測沒有明顯影響;同時可以看出,開始加料時體系中小微粒的計數增長得非常快,這說明了洗滌硫酸液體和有機相的界面處存在相當大的過飽和度和較強的攪拌擾動,使得硫酸鈣晶核迅速大量形成,在洗滌過程中晶體的成核誘導期非常短,直接進入成核階段;同時一開始形成的少量較大微粒(弦長50~1 000 μm)的消失,說明了在結晶過程中有局部微粒的破碎和溶解;在0~8 min硫酸鈣晶體的成核和生長基本完成,此后基本進入體系中殘留過飽和度消耗階段,此時體系過飽和度已經大大降低,不再生成新的晶核,弦長<10 μm和10~50 μm的微粒計數部分下降,較大顆粒(>50 μm)的計數開始上升,同時實驗觀察到有機相的透明度增加,部分微粒聚并成為較大的無法檢測的結塊聚沉。圖7為洗滌過程中不同時刻弦長分布(CLD)。由圖7可知,隨著洗滌時間的增加,微粒的計數和弦長分布呈現了由小到大再逐漸變小的規律,說明了硫酸鈣晶體的形成基本遵循成核、生長、體系中過飽和度消耗和結塊沉降的過程。
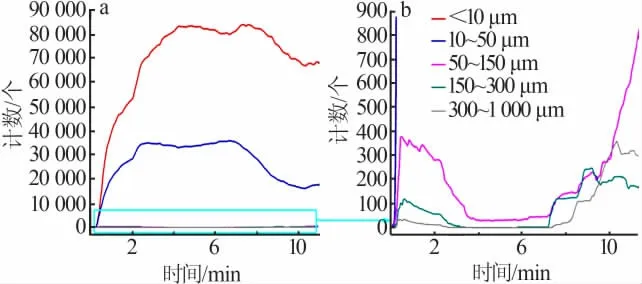
圖6 洗滌過程中微粒計數變化Fig.6 Counts of particles vary with time in the scrubbing process
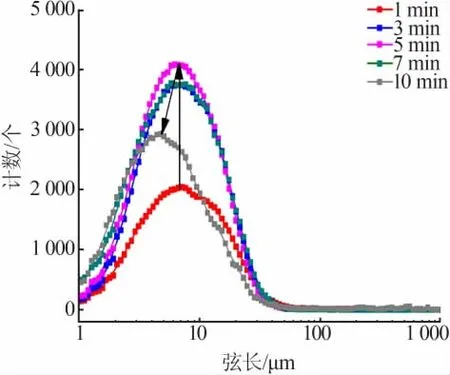
圖7 洗滌過程中微粒弦長分布(CLD)—時間變化Fig.7 Chord length distribution(CLD)varies with time in the scrubbing process
圖8 和圖9分別為洗滌時間為10 min時,上述過程所得洗滌石膏的SEM照片與XRD譜圖。由圖8可以看出,洗滌石膏主要由最大長度為20 μm的硫酸鈣晶體搭接構成;由圖9可見,洗滌10 min時,所得石膏的物相組成為CaSO4·0.5H2O。

圖8 洗滌硫酸鈣的SEM照片Fig.8 SEM images of CaSO4·nH2O in the scrubbing process

圖9 洗滌硫酸鈣的X射線衍射譜圖Fig.9 XRD pattern of CaSO4·nH2O in the scrubbing process
2.2.2 洗滌有機相磷酸反萃取實驗結果
對上述洗滌條件下所得有機相用去離子水進行反萃,仍按照式(4)對整個洗滌-反萃過程中磷酸的反萃取率進行計算,結果見表6。

表6 洗滌-反萃過程實驗結果Table 6 Results of H3PO4 stripping from the scrubbed organic phase
表6 結果說明,在20℃時對洗滌后的有機相進行反萃,當反萃取相比V(A)/V(O)為1∶3、2∶3和1∶1時,H3PO4的反萃取率分別為59.29%、68.73%和70.83%。而對比表中5中a1~a3組的實驗結果,發現在相同的反萃取溫度和相比V(A)/V(O)下,采用H2SO4溶液對有機相直接進行反萃取時,H3PO4的反萃取率分別為56.33%、85.92%、92.28%。顯然,在較高的反萃取相比[V(A)/V(O)為1∶1]時,采用洗滌-反萃流程,H3PO4的反萃取率降低了20%左右,這部分H3PO4反萃取率的損失應該為洗滌過程中有機相中的部分H3PO4進入洗滌水相被帶走所致,而實際流程中可將洗水返回萃取工段以回收其中的H3PO4;而從磷鈣比[n(H3PO4)/n(Ca2+)]來看,洗滌反萃酸中的磷鈣比均大于3 000,且隨著反萃取水相用量的增大,磷鈣比并沒有減小,這是因為分離洗滌石膏固相后消除了CaSO4·nH2O溶解平衡對反萃酸品質的影響。對比表5中a1~a3組的實驗結果發現,通過洗滌-反萃流程得到的反萃酸,其磷鈣比增大了65倍以上,與直接采用H2SO4溶液對有機相中的H3PO4進行反萃相比,采用先以H2SO4進行洗滌有機相分離硫酸鈣石膏后再以軟水反萃取H3PO4的流程,可顯著降低反萃取磷酸中Ca2+含量。
2.3 洗滌石膏的綜合利用分析
常見的硫酸法濕法磷酸生產工藝按照其所得的水合硫酸鈣結晶類型分為二水物法、半水物法和再結晶法(半水-二水法和二水-半水法)。以本實驗用磷礦(表2)為例,采用二水法生產工藝(80℃,攪拌轉速為400 r/min,攪拌時間為4.5 h,反應料漿液固質量比為3∶1,制備磷酸質量分數為21.32%(P2O5),磷礦粉過0.178 mm篩),實驗室制備的磷石膏SEM、EDS、XRD圖分別見圖10、11。
圖10 表明,以二水物法工藝處理本實驗所用磷礦所得磷石膏的主要物相成分為CaSO4·2H2O和SiO2;圖11表明,硫酸鈣固相為典型的斜方六面板狀和棱柱狀晶體,面掃圖像表明磷石膏中混合著較大量的不規則的酸不溶SiO2。而SiO2作為硫酸法石膏中的主要固體雜質,對磷石膏的綜合利用會產生不利的影響,比如在磷石膏制硫酸聯產水泥工藝中,磷石膏中SiO2含量對磷石膏熱分解生產工藝和產品質量影響較大,采用高硅含量的磷石膏時熱分解過程中易結圈,水泥熟料產品質量不穩定;在磷石膏制備硫酸銨和碳酸鈣的過程中,磷石膏中的高SiO2含量會導致其副產的碳酸鈣產品質量不合格,不能作為市場產品銷售從而降低了磷石膏綜合利用的可行性,增大了磷石膏處理的難度等[19-20]。本文的研究結果表明,采用鹽酸法工藝流程時得到的石膏屬于清潔白石膏,不含SiO2等固體雜質,這就很大程度上降低了工藝中石膏的處理難度,提升了其綜合利用價值和潛力。

圖10 四川會東磷礦磷石膏的X射線衍射譜圖Fig.10 XRD pattern of phosphogypsum generated by phosphate rock in Huidong,Sichuan

圖11 四川會東磷礦磷石膏的掃描電鏡/能譜面掃圖像Fig.11 SEM and EDS mapping images of phosphogypsum generated by phosphate rock in Huidong,Sichuan
從產生石膏的量來看,以傳統二水硫酸法工藝為例,100份質量的磷礦完全被H2SO4分解時的石膏值可按照下式計算:
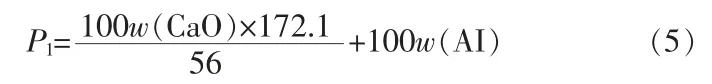
式中:w(CaO)為磷礦中CaO質量分數,%;w(AI)為磷礦中酸不溶物質量分數,%;172.1為CaSO4·2H2O摩爾質量,g/mol;56為CaO摩爾質量,g/mol。
對于本實驗中所用磷礦(表2),若以SiO2含量近似代表總酸不溶物含量AI,其100份質量磷礦產生的干基磷石膏計算值約為137份,其中主要成分為CaSO4·2H2O和SiO2。如按照本文中的鹽酸法工藝,假設被萃取入有機相中的CaCl2全部以CaSO4·2H2O的形式成為洗滌石膏,則此工藝中的理論石膏值:

式中:ECaCl2為萃取過程中CaCl2的萃取率。
之前的實驗工作表明,通過萃取劑(80%TBP+20%煤油,體積分數)逆流萃取上述磷礦-鹽酸浸出液中的H3PO4,經四級逆流萃取后CaCl2的共萃取率在10.27%左右,按照此值計算所得的洗滌石膏值P2約為11.9份,僅為二水硫酸法理論石膏值的8.7%左右。可見在工業副產鹽酸來源有保障、CaCl2鹽水后處理技術相對成熟的情況下,鹽酸法路線在節約硫資源消耗和減少磷石膏排放方面可以展現出其巨大的優勢。
3 結論
1)采用H2SO4溶液一步沉淀有機相中的CaCl2并反萃取H3PO4時,提高反萃取劑的體積用量和反萃取溫度可增大H3PO4的反萃取率;60℃時,當反萃取的相比V(A)/V(O)=1∶3時,H3PO4反萃取率可達75%以上,相比V(A)/V(O)增大到1∶1時,H3PO4反萃取率可達96%以上。硫酸鈣沉淀的晶體尺寸主要受到H2SO4濃度的影響,H2SO4濃度降低,硫酸鈣晶體向長條板狀過度,其長徑比增大明顯,同時會出現細小碎晶,過低的H2SO4濃度不利于硫酸鈣的結晶和過濾質量的提高。由于硫酸鈣在反萃酸中存在溶解平衡,因此反萃磷酸中的CaCl2質量分數始終略高于0.2%,此法對反萃磷酸的凈化效果有限。
2)洗滌反萃兩步法流程實驗的結果表明,在20℃,相比V(A)/V(O)分別為1∶3、2∶3和1∶1,以軟水對洗滌后有機相中的H3PO4進行反萃取,H3PO4的反萃取率分別為59.29%、68.73%和70.83%,有機相中H3PO4的洗滌損失率為20%左右,但此時反萃酸中磷鈣比n(H3PO4)/n(Ca2+)均>3 000,約為用H2SO4直接反萃時的65倍以上,反萃酸中Ca2+的去除率顯著提高。聚焦光反射測量技術對洗滌過程的檢測表明,實驗條件下H2SO4液滴可在有機相中良好分散,洗滌過程在10 min內基本完成,生成的石膏均為CaSO4·0.5H2O。
3)本實驗中所得硫酸鈣均為不含SiO2的清潔石膏,與硫酸法工藝中產生的二水石膏相比,其后處理難度小,綜合利用潛力大,且其石膏產量僅為二水硫酸法理論石膏產量的8.7%左右,顯著降低了濕法磷酸過程中的石膏處理量。
猜你喜歡
作文·小學低年級(2025年2期)2025-02-13 00:00:00
小雪花·小學生快樂作文(2024年11期)2024-12-31 00:00:00
作文·小學低年級(2024年2期)2024-04-29 00:00:00
作文·小學低年級(2023年3期)2023-04-29 00:00:00
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
小主人報(2022年4期)2022-08-09 08:52:06
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
