以肝硬化合并急性腹痛為表現的紅細胞生成性原卟啉病1例
2021-07-13 08:23:40劉曉燕蘇海濱
傳染病信息 2021年3期
李 會,李 晨,劉曉燕,蘇海濱
1 病例報告
1.1 病史 患者,男,40歲,已婚,山西人,煤礦辦公室工作人員,因“間斷乏力、身目黃染2年余,加重伴腹痛6 d”于2018年7月4日入住中國人民解放軍總醫院第五醫學中心。患者2015年11月出現乏力,身目黃染,尿黃如豆油色,后逐漸加重,2016年2月于我中心就診,診斷為“肝硬化失代償期合并腹水(肝硬化原因不明)”,予對癥保肝等治療后癥狀好轉出院。2018年3月患者再次出現上述癥狀,自行口服“中藥(具體不詳)”治療2月余,無效。6月27日上述癥狀加重,并伴有腹痛,疼痛部位不固定,有時放射至右側胸部及后腰部,持續性鈍痛、針扎樣痛、絞痛、燒灼樣疼痛交替,無明顯緩解及加重因素。6月30日再次就診于我中心,查肝功能異常,遂以“肝硬化失代償期”收入病房。患者既往約10歲時出現“皮膚病(自訴為日光過敏性皮炎,表現為暴露部位的燒灼樣感、紅腫、水泡、結痂)”,間斷口服“中藥、西藥”等治療,效果不佳,仍反復發作,患者哥哥及其女兒也有類似表現(較患者表現輕微),父母非近親結婚,無其他基礎疾病及遺傳病診斷史,無煙酒史及吸毒史,育有1女1子,1子為“白化病”患兒,女兒、愛人及父母均身體健康。
1.2 入院查體 生命體征平穩,營養中等,神志清楚,精神差,面色晦暗,皮膚、鞏膜輕度黃染,口周及雙上肢可見片狀結痂性皮膚(光過敏皮膚損傷),肝掌陽性,蜘蛛痣陰性。全身淺表淋巴結未觸及腫大。心肺未見異常。腹部平軟,未見腹壁靜脈曲張,右下腹壓痛明顯,輕微反跳痛,肝右肋下2 cm可觸及,緣鈍,質中,無觸痛,莫菲氏征陰性,脾左肋下未觸及。移動性濁音可疑陽性,腸鳴音正常。雙下肢無水腫。神經系統查體未見異常。
1.3 實驗室檢查及其他輔助檢查 腹水常規未提示感染(腹水常規:顏色黃、透明度清、李瓦它試驗陰性、細胞總數1146×106/L、WBC 146×106/L、中性粒細胞百分比23%、間皮細胞百分比8%、淋巴細胞百分比69%),CRP 33.5 mg/L,降鈣素原1.08 ng/ml。肝功 能:ALB 40 g/L、T/DBIL 201.3/158.8 μmol/L、ALT 216 U/L、AST 458 U/L、ALP 233 U/L、谷氨酰轉肽酶(γ-glutamyl transpeptadase, GGT)584 U/L,γ- 球蛋白 19.5%,IgG 18.39 g/L,銅蘭蛋白0.43 g/L,鐵46.7 μmol/L。凝血功能:凝血酶原活動度(prothrombin time activity, PTA)58.9%、國際標準化比值(international normalized ratio, INR)1.22。血常規:WBC 5.12×109/L、HGB 127.00 g/L、中性粒細胞百分比75.6%、PLT 25.00×109/L。甲狀腺功能正常。甲、乙、丙、戊型肝炎病毒標志物陰性,單純皰疹病毒、EB病毒、巨細胞病毒相關血清學抗體陰性。抗核抗體及肝病自身抗體均陰性。腹部增強CT:①肝硬化、脾大、副脾、腹水、食管及胃底靜脈曲張、脾靜脈曲張。②膽囊炎。③肝囊腫,右腎囊腫。④肝動脈變異。肺部CT:左肺上葉陳舊性病變。腹部立位平片:不完全腸梗阻,可見氣液平面。肝臟血管超聲未見異常。泌尿系統超聲未見異常。盆腔CT:盆腔積液、腹腔積液、盆腔鈣化灶。
1.4 診治經過 入院后結合患者病史、查體及輔助檢查,排除病毒性、自身免疫性、酒精性等常見肝損傷病因,并查閱了相關遺傳性代謝性肝損傷的文獻[1-2],排除肝臟血管病變、肝豆狀核變性、鐵代謝異常血色病及抗胰蛋白酶缺乏癥等先天性遺傳代謝性肝損傷疾病。肝硬化原因有待進一步明確,腹痛原因為腸梗阻及腹膜炎(臨床診斷),并考慮腸梗阻為腹痛的主要原因。治療上給予禁食水、胃腸減壓、石蠟油灌腸、抗感染、保肝、降酶、退黃、抗淤膽等內科綜合治療,效果不佳,患者腹痛無明顯緩解,疼痛范圍逐漸擴展至腰背部、膀胱區、前胸、后背、上臂及大腿,疼痛性質同前,肝功能及凝血功能進行性惡化,進展為肝衰竭。7月18日復查肝功能:ALB 30 g/L、T/DBIL 437.6/324.3 μmol/L、ALT 251 U/L、AST 480 U/L、ALP 216 U/L、GGT 271 U/L。凝血功能:PTA 38.8%、INR 1.60。血常規:WBC 6.12 ×109/L、HGB 90.00 g/L、中性粒細胞百分比82.8%、PLT 36.00×109/L。再次重新分析病情,結合患者既往有日光性皮炎、哥哥及其女兒有類似皮膚病,此次發病主要以肝功能異常、腹痛、腸梗阻為主要臨床表現,抗生素及胃腸減壓等治療無效,查體見口周破潰,呈放射狀萎縮性紋理(圖1),雙手部皮膚粗糙(圖2),考慮卟啉病。
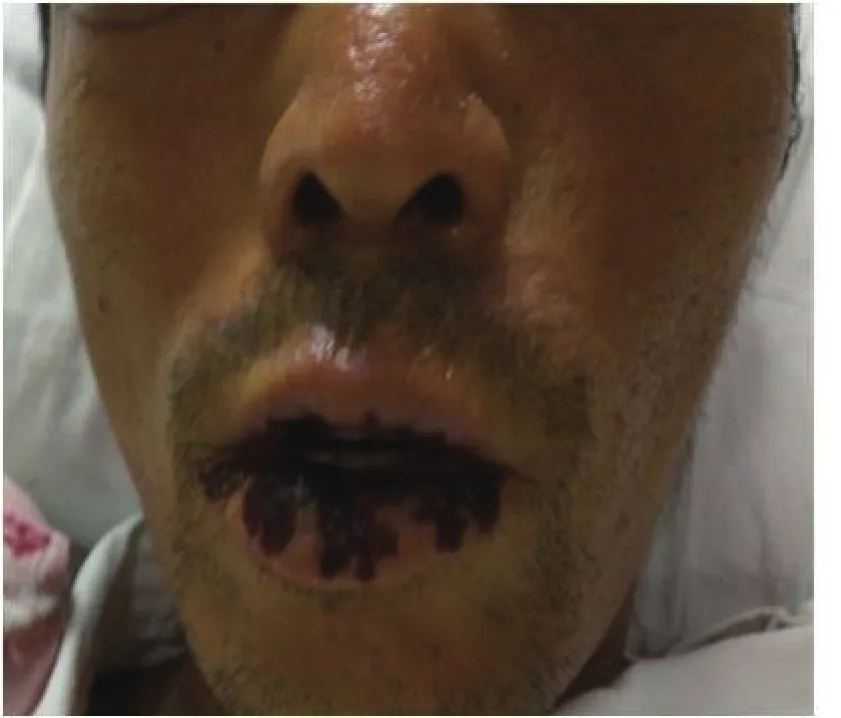
圖1 患者口唇部皮膚表現口周破潰,呈放射狀萎縮性紋理Figure 1 Skin manifestations on patient’s lip and mouth

圖2 患者手部皮膚表現橈側及虎口處皮膚增厚,手背皮膚粗糙Figure 2 Skin manifestations on patient’s hand
1.5 確診 2018年7月20日北京協和醫院尿卟啉檢測結果為陰性,紅細胞內游離原卟啉的檢測結果:細胞內鋅卟啉為57.5 μg/gHb(參考值為0~4.7 μg/gHb)。患者尿液放置曝光以及在WOOD燈下無顏色變化。血液基因學檢測:常染色體隱性紅細胞生成性原卟啉病(erythropoietic protoporphria,EPP)相關基因亞鐵螯合酶外顯子及內含子區域存在3處雜合突變,即 c.892C>T(胞嘧啶>胸腺嘧啶)(圖3A),c.68-23C>T(胞嘧啶>胸腺嘧啶)(圖3B),c.315-48T>C(胸腺嘧啶>胞嘧啶)(圖3C)。最終明確診斷:EPP。7月21日患者自動出院,于當天死亡。
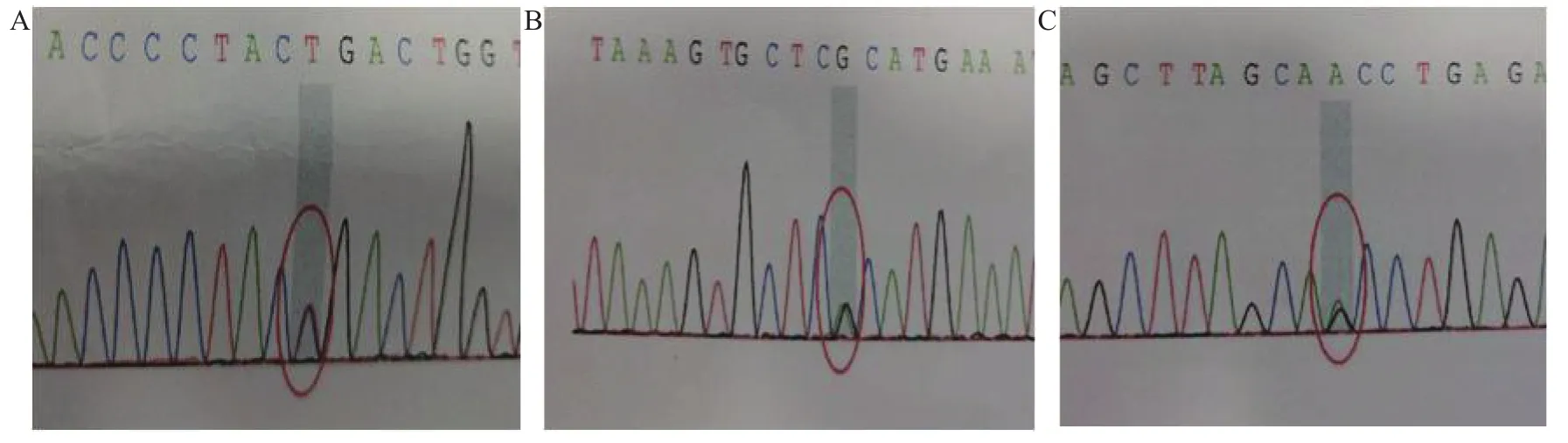
圖3 患者亞鐵螯合酶基因突變點檢測結果A.c.892C>T(胞嘧啶>胸腺嘧啶);B.c.68-23C>T(胞嘧啶>胸腺嘧啶);C.c.315-48T>C(胸腺嘧啶>胞嘧啶 )Figure 3 Detection results of FECH gene mutation points in patient
2 討 論
卟啉病是在血紅素生物合成途徑中,因某些酶的缺乏或異常,引起未轉化為血紅素的卟啉及(或)卟啉前體(如δ-氨基-γ-酮戊酸和膽色素原)在體內過度分泌及蓄積,并在組織中沉積,最終由尿和糞便排出的一組遺傳代謝性疾病。血紅素在哺乳動物組織中通過受酶調控的8個步驟進行合成,根據血紅素生成步驟中所需酶的不同,將卟啉病分為8個類型,每一類型均與血紅素合成通路上的一種酶缺陷有關,并有相應的基因突變位點,當基因位點發生突變后可形成不同類型的卟啉病,表現為卟啉、卟啉前體或者兩者特征性的蓄積和異常分泌。各類型卟啉病有其特異的相關酶缺乏的病理生理機制,基因學檢測出相應的酶學基因突變點為診斷卟啉病的最直接證據。遺傳方式可能為常染色體顯性或隱性遺傳。
EPP是因血紅素合成最后一步中的催化酶亞鐵螯合酶活性缺陷或活性低下,使體內原卟啉IX水平升高并在體內蓄積沉積于皮膚等全身組織所致的疼痛性遺傳性皮膚病[3]。大部分EPP是由于亞鐵螯合酶一個等位基因突變所致的具有不同外顯率的常染色體顯性遺傳病。亞鐵螯合酶基因已被定位于染色體18q21.3-22區域,含有11個外顯子和10個內含子,少數患者(占4%)以常染色體隱性方式遺傳,常染色體隱性遺傳是發生肝衰竭的危險因素[4]。因染色體突變所致亞鐵螯合酶缺乏,從而使原卟啉在紅細胞、肝臟、皮膚中過度沉積而致病。原卟啉是一種由肝臟分泌的親脂性分子,因此EPP患者有膽石癥和膽道梗阻性發作的風險,并具有較高的肝衰竭風險,為EPP患者發病率和病死率較高的主要原因[5]。
本例患者基因學檢測亞鐵螯合酶存在3處雜合突變,突變位點c.892C>T(胞嘧啶> 胸腺嘧啶)為致病突變,導致蛋白翻譯終止;突變位點c.68-23C>T(胞嘧啶>胸腺嘧啶)為可疑致病突變,影響蛋白剪切;突變位點c.315-48T>C(胸腺嘧啶>胞嘧啶)為疾病相關多態性位點,常常和明確致病突變點組合成復合雜合突變,從而導致EPP[6-7]。此3處基因突變點分別位于兩條染色體上,且每條染色體上均有一處突變為致病性突變,結合患者病史特點及細胞內鋅卟啉明顯升高,故可以明確診斷EPP。但在診斷上,尚存在不足之處:①該病例因患方原因未進一步明確患者父母及子女的基因學檢測以充分驗證該診斷,同時未對該患者進行肝穿刺病理活檢以明確肝臟病理;②在鑒別診斷中,未進行充分的基因檢測和進一步檢查以明確排除抗胰蛋白酶缺乏癥和藥物性肝損傷的可能;③在診療過程中,未能及時準確地明確肝硬化的病因診斷,喪失最佳治療時機,加之花費較高,導致患者最終死亡。
EPP多在兒童期發病,比較罕見,也可遲至成年發病,男性多于女性,發病率波動在1/75萬~1/20萬[8]。EPP的臨床表現主要有以下幾個方面:①光敏性:為無水泡的急性疼痛性光敏反應,兒童時期或更早時期發病,從第一次照射陽光開始,持續終生,最初表現為陽光照射下暴露區皮膚刺痛、紅斑、水腫及灼傷,持續數小時至數天,與陽光照射的強度和持續時間有關,反復的光敏性發作導致皮膚出現蠟質或皮革狀增厚及角化過度外觀,嘴唇呈線萎形縮性皺紋和偽橫紋。其主要機制為原卟啉在血液循環及皮膚中沉積,吸收光能后呈激發狀態,與氧結合生成活性氧,導致膜蛋白的交聯、膜脂質的過氧化,從而使細胞膜和線粒體成為原卟啉誘導損傷的主要靶點,而整個反應過程中肥大細胞釋放的5-羥色胺及花生四烯酸促進光反應的進行。②腹痛:主要表現為發作性的絞痛、脹痛、針扎樣和/或刀割樣交替疼痛,疼痛局限或放射至腰背部,伴有惡心嘔吐、頑固性便秘甚至腸梗阻,應用止痛藥有效。其發病機制可能為胃腸組織內5-羥色胺增加、胃腸自主神經功能紊亂、原卟啉毒性作用直接刺激胃腸道平滑肌。本例患者有明顯腹痛、腸梗阻表現,并且有放射至膀胱區、前胸、后背部、上臂及大腿部燒灼樣的疼痛,常易被誤診為“腹痛原因待查”,該患者在診療過程中也考慮腸梗阻及腹膜炎為腹痛原因,但經積極抗感染及解除腸梗阻治療后,疼痛并未得以緩解。③肝損傷:大約5%~20%的EPP患者出現肝損傷表現[9]。國內外文獻均有相關肝膽系統受累的EPP患者的報道,并且均發生了嚴重的肝損傷,甚至肝衰竭[10-14]。還有研究表明較高的紅細胞原卟啉水平是EPP患者病情嚴重程度和肝損傷加重的主要決定因素[15]。其發病機制為原卟啉沉積于肝細胞、巨噬細胞、庫弗細胞和膽管系統,在偏差顯微鏡下呈雙折射性的晶狀體,過多的原卟啉在肝臟分泌入膽汁過程中,沉積于肝細胞及膽管腔內并聚集成小晶狀體,從而阻塞膽道并損傷肝細胞,而且原卟啉在肝臟淤積后易形成膽結石。原卟啉具有直接的肝毒性,當原卟啉生成過多超過肝臟代謝時,在肝臟的長久蓄積可誘發肝損傷和肝硬化。另外,其他類型的原卟啉病經常有合并神經精神癥狀,可有周圍神經、自主神經、中樞神經系統功能及精神狀態的損害,其發病機制目前尚未明確,可能與卟啉前體物質破壞神經組織有關,并未發現與原卟啉相關,所以臨床上EPP患者的神經精神癥狀并不常見。
目前尚無根治EPP的有效治療方案,以降低光敏感性、減少原卟啉產生、促進原卟啉排出為主,輔以其他對癥支持治療緩解癥狀。本病的基礎治療是控制光敏感,避免光損傷,口服胡蘿卜素降低光敏性,歐洲聯盟批準光損傷保護劑Afamelanotide用于治療EPP患者的皮膚損傷[16-17]。口服鵝脫氧膽酸能降低體內原卟啉水平。考來烯胺可阻斷原卟啉的腸肝循環,促進原卟啉從糞便中排出。熊去氧膽酸也可用于治療EPP[18]。還有研究者采用反復輸血提供血紅素,血漿置換清除過多的原卟啉來治療EPP合并急性膽汁淤積[19]。因肝移植并不能改變患者基因變異所致的酶缺陷,移植后仍可出現過多的原卟啉沉積于肝臟,造成肝損傷,因此肝移植并不作為推薦。肝和骨髓聯合移植應被認為是一種適合嚴重肝受累的EPP病例的治療方法[8]。骨髓或造血干細胞移植、干細胞基因治療有待進一步深入研究[20]。
EPP是一種罕見的遺傳代謝性疾病,臨床表現形式有多樣性、間歇性、反復性、漸進性、隱匿性特點,首診形式多樣,在皮膚科、普外科、神經科、消化科、血液科、兒科、肝病科均有病例報道。本例患者以肝損傷病因不明首診,在診療初期皮膚損傷癥狀被忽視或被認為與此病無關;在病情進展階段,并發了急性腹痛,以積極對癥治療腹痛(腹膜炎及腸梗阻)后,肝功能仍在持續惡化;在重新系統篩查肝損傷病因,結合皮膚損傷表現、腹痛特點及查閱相關文獻后才考慮為EPP,臨床診療過程曲折,極易造成疾病的漏診、誤診。
綜上所述,通過本例EPP的診療,豐富了該病的診療經驗,充分意識到只有對卟啉病可能引起的皮膚損傷、腹痛、肝硬化甚至肝衰竭有所認識,形成將皮膚損傷、腹痛、肝損傷進行“一元論”的臨床思維,才能做出正確的臨床判斷,選擇合理的臨床輔助檢查手段,達到早期診斷及治療,從而提高臨床醫生對卟啉病的認識和診療水平。
