關于遺傳基因檢測中基因變異臨床意義分級的建議
2021-07-09 05:18:56天津市醫學會醫學遺傳學分會天津市醫學會遺傳咨詢分會
天津醫藥 2021年6期
天津市醫學會醫學遺傳學分會,天津市醫學會遺傳咨詢分會
由于近年來高通量測序技術,尤其是全外顯子組測序技術的廣泛應用,使得對海量的基因變異進行分析解讀成為可能。美國醫學遺傳學與基因組學學 會(American College of Medical Genetics and Genomics,ACMG)發表了基因變異致病性的分類指南,包括非腫瘤性遺傳病相關的胚系變異(為主體)的分類指南[1-3]、拷貝數變異的分類指南[4-7]、線粒體tRNA(mitochondrial tRNA)基因變異的分類建議[8-9]。近年關于致病性分類方法還在不斷修訂中[10-15]。在臨床實踐過程中,對于遺傳基因檢測報告的解讀經常會產生誤解,一方面源自變異水平的致病性分類方法尚不夠完善;另一方面源自臨床醫生與實驗室人員理解上的偏差。實驗室專家往往強調基因變異的分類僅僅與變異本身的性質相關,而與具體案例(患者、攜帶者或者胎兒)的表型無關,而臨床醫生則需要直接面對臨床案例,不僅要考慮變異本身的性質,而且要考慮案例的具體情況。
為了彌補實驗室人員與臨床醫生之間關注焦點的差異,降低臨床醫生理解檢測報告的難度,本文建議:基因檢測實驗室不僅要對變異的致病性進行分類,還應通過與臨床科室的密切溝通,對疑似基因相關表型譜與具體臨床案例之間的相關程度進行評估,并在此基礎上對基因變異進行臨床意義的分級。通過在檢測報告中同時給出基因變異的致病性分類以及臨床意義的分級,有助于臨床醫生準確地理解檢測報告,也有助于下一步準確地向患者和(或)家屬提供臨床遺傳咨詢服務。
1 臨床信息采集與疑似致病基因相關表型的回顧
1.1 詳細表型和家族史采集的重要性 臨床案例的詳細表型和家族史對于后續基因檢測結果的分析與解讀具有極為重要的意義。表型資料與家族史的準確度和全面性不僅可能影響醫生能否選擇出最合理的檢測項目,還可能影響到實驗室在數據分析解讀中能否定位到真正需要被關注的基因,也會影響到能否準確地進行致病性的評估。在致病性的評估過程中,一些證據點的評價需要考慮到臨床相符的程度,如PP1(符合基因型-臨床表型的共分離)和PP4(臨床表型相符并且高度特異)[1]。因此,臨床表型和家族史的采集是否完整與準確可能會影響最終的醫療實踐。
在完成了基因檢測之后,還可以根據基因檢測報告所提示的疑似診斷再次進行表型采集,以便發現較為次要的表型,進一步確認或者排除診斷。尤其是針對攜帶可疑基因變異的家庭成員,也應盡量采集其表型,幫助對基因型-表型之間的相關性進行評估及確認。
1.2 輔助進行表型描述以及查詢的數據庫和網站
1.2.1 人類表型標準用語聯盟(Human Phenotype Ontology,HPO)和中文人類表型標準用語聯盟(CHPO)表型標準化數據庫 隨著對人類疾病研究的逐漸深入,科研工作者們越來越意識到臨床表型數據的重要性,對基因型與表型數據進行聯合分析成為眾多疾病研究的一個方向。HPO旨在提供人類疾病中用于描述表型異常的標準詞匯,目前包含約11 000個名詞和超過115 000條關于遺傳性疾病的注釋,還提供了一套針對約4 000種疾病的注釋(https://hpo.jax.org/app)[16-17]。HPO中每個術語描述了一種異常表型,如房間隔缺損。HPO建立者從醫學文獻、Orphanet、利用染色體組分資源建立的人類染色體不平衡與表型數據庫(DatabasE of genomiCvarIation and Phenotype in Humans using Ensembl Resources,DECIPHER)和在線人類孟德爾遺傳(Online Mendelian Inheritance in Man,OMIM)數據庫獲取信息并進一步開發,持續進行詞條的維護和完善。HPO的建立有助于臨床醫生以標準化的醫學術語來描述罕見病患者的表型,這樣不僅有利于診斷疾病,確定致病基因,還能幫助研究人員尋找疾病與特定表型之間的關系。2016年初,國內相關領域的專家共同成立了CHPO,基于HPO網站進行詞匯的中文翻譯和編輯優化,并建立了CHPO搜索引擎(http://www.chinahpo.org),提供詞庫的免費下載,獲得了國內同行的廣泛關注和贊許。
1.2.2 OMIM數據庫 OMIM數據庫(http://www.omim.org)是關于人類基因和遺傳紊亂的數據庫,通過對新的病癥分類并命名、收錄表型和相關致病基因的關系來收錄人類孟德爾疾病信息,可以通過臨床特征、表型和基因型來搜索對應的信息。OMIM數據庫包括大量已知的遺傳病、遺傳決定的性狀及其基因,除了簡略描述各種疾病的臨床特征、診斷、鑒別診斷、治療與預防外,還提供已知有關致病基因的連鎖關系、染色體定位、組成結構和功能、動物模型等資料,并附有經縝密篩選的相關參考文獻。OMIM數據庫持續更新,是用來查詢特定致病基因相關臨床表型的較為可靠的數據庫[18]。
1.2.3 GeneReviews數據庫 GeneReviews數據庫(https://www.ncbi.nlm.nih.gov/books/-NBK1116)是美國國立衛生研究院資助,由美國華盛頓大學組織編撰并維護的一系列在線叢書數據庫。該叢書的編寫目的是為繁忙的一線醫學工作者提供可以直接在臨床上應用的遺傳病相關信息。GeneReviews內容非常詳實,并支持遺傳病外顯率的查詢。不僅如此,其還提供包括鑒別診斷、治療方案、風險因素、監控方案和遺傳咨詢等信息,適合臨床醫生和遺傳咨詢師使用。截至2021年4月25日,GeneReviews共收錄了796個章節且在不斷增加中。2016年底,國內專家開始著手建立中文版GeneReviews(https://genereviews.nrdrs.org.cn/paper/index),翻譯全部現有詞條并根據英文原版及時進行更新,添加了更多英文版未涵蓋的詞條和中國人群特有的信息,如中國人群中的發病率和變異熱點等。
1.2.4 人類基因突變數據庫(Human Gene Mutation Database,HGMD) HGMD(http://www.hgmd.cf.ac.uk)存儲了與人類疾病相關的致病變異信息,內容動態更新,不斷添加新的數據,并且淘汰被認為可能存在錯誤的信息[19]。該數據庫已經覆蓋了超過1.1萬個可能與人類疾病相關的基因,以及超過29萬種有關的變異(以致病性的變異為主),并且注明可能與每個變異相關的臨床表型。該數據庫已經成為臨床基因測序結果的解釋中不可或缺的重要工具之一。
1.2.5 MalaCards數據庫 MalaCards數據庫(http://www.malacards.org)是人類疾病及其注釋的綜合匯編[20-21]。當前版本包括來自74個來源的21 369種疾病的信息。對于每一種疾病,數據庫都會顯示一張帶有關于該疾病的各種注釋信息的“疾病卡”,匯總了該疾病的各種已知信息。這些信息來自GeneCards數據庫、文獻檢索和GeneAnalytics基因集分析工具。MalaCards數據庫使用一個自動計算信息檢索引擎,通過利用遠程數據以及GeneCards平臺收集的信息,整理和填充疾病卡。MalaCards數據庫整合了專門疾病和一般疾病列表,包括罕見疾病、遺傳病和復雜疾病等。MalaCards中每種疾病的相關基因的平均數多于OMIM,其特點是大而全,注解來源于文獻,但可靠性不易判斷。當在OMIM上未發現與基因對應的疾病時,可嘗試該數據庫。
1.2.6 DECIPHER數據庫 DECIPHER數據庫(https://decipher.sanger.ac.uk/index)是目前分子遺傳學中最重要的生物信息學數據庫之一。DECIPHER是一項國際化的合作項目,截至2021年4月25日,向該數據庫上傳數據的研究項目達到294個,開源的病例記錄超過3.85萬個。用戶可以通過檢索數據庫發現一系列相關的遺傳疾病信息,包括變異位點和臨床表型等,從而提高臨床診斷效能。
1.2.7 人類基因組結構變異(database of genomic variants,DGV)數據庫 DGV數據庫(http://dgv.tcag.ca/dgv/app/home)是正常人群的染色體結構變異數據庫,提供了人類染色體結構變異的概況信息,記錄了一系列基因變異與表型相關的信息,是目前在結構變異的致病性評級中常用的對照人群數據庫。DGV數據庫收錄了健康人群樣本中大于50 bp的基因組結構變化信息[22]。
1.2.8 MitoMap(人類線粒體基因組)數據庫 MitoMap數據庫(https://mitomap.org/MITOMAP)[23-24]收錄了大量人類線粒體的全長序列數據(>51 800份)以及超過1.9萬個基因變異位點,并對其中可能具有致病性的變異位點相關的臨床表型進行了羅列。該數據庫是臨床線粒體病測序實驗室最常用的數據庫之一。
1.2.9 Phenolyzer工具 Phenolyzer工具(http://phenolyzer.wglab.org)可以十分方便地提供從標準化表型到可能相關基因的檢索[25],目前已經被廣泛應用于遺傳分析工作中。該工具不僅能給出與特定表型(或一組表型)相關的基因信息,并且給出了按照優先級從高到低的排序。
2 基因變異致病性分類
2.1 基本的五分類 根據ACMG的相關指南,不論何種類型的遺傳物質變異(點突變/拷貝數變異/線粒體變異),都可以按照五分類法對變異的致病性進行分類:致病性變異(pathogenic variants)、可疑致病性變異(likely pathogenic variants)、致病性不明確的變異(variants of unknown significance)、可能良性的變異(likely benign variants)、良 性 的 變 異(benign variants)[1]。
2.2 對致病性變異的細分 在解讀致病性變異和可疑致病性變異時,還應當適時地考慮到外顯率、表現度,甚至還需要考慮到致病機制。同一個致病基因的不同變異可能具有迥異的外顯率和表現度,甚至還可以引起完全不同的表型。這些方面都是在基因檢測結果的臨床解釋中要考慮到的。尤其是當患者家庭中存在變異攜帶狀態符合患者的特點而沒有臨床表型的家庭成員時,對于外顯率較低或者是極低的變異,應當盡量在臨床報告中給出相關的信息。即使根據基因變異分類指南可以將此類變異分類為“致病性變異”,也推薦使用“外顯率較低的致病性變異位點”、“外顯率極低的致病性變異位點”或者“患病風險相關的基因變異”對此類變異進行描述。
對基因變異進行致病性分類時,還常常遇到一種窘境:由于臨床信息可能不全面,或者不能確認患者與送檢父母之間是否為親生關系,使得本來可以獲得較高級別的證據強度卻不得不調低,僅能將變異分類為“致病性不明確的變異”,而在采集到完整的信息之后,恰好能根據更新的證據將致病性分類上升為“可疑致病性變異”。遇到此類情形時,推薦將此類變異的分類描述為“尚不能肯定具有致病性(variants of unknown significance,VUS),但偏向于具有致病性的變異(VUS-likely pathogenic,VUS-LP)”。
2.3 基因變異分類的擴展 針對可能與臨床相關的疾病易感性變異位點而言,原則上應該綜合是否已有功能學研究的證據支持、相關疾病(或特定表型)的遺傳度、風險相關性的強度等因素。風險相關的強度可用比值比(OR值)、相對危險度(RR值)、加權遺傳風險(CGR值)等指標來描述。本文建議,可根據是否已有功能學驗證的證據將變異位點分為兩類:一類位點不僅有群體統計數據支持,而且所在的基因與所關注的疾病之間的關系已經有充分的功能研究證據;另一類位點則僅僅有群體統計數據支持,但是尚缺乏可靠的功能研究證據支持。
尚有一類基因變異,它們可能有較高的人群頻率,通常并不引起疾病,但是有可能會引起特殊的生化表型或者特殊的體征。此類變異可以劃分為功能多態性變異的范疇。在特定的情況下,檢測到此類變異也可能輔助臨床解釋。因此,在基因變異的分類中,除了按是否具有致病性進行劃分以外,補充功能多態性變異也有助于臨床實踐和遺傳咨詢。見表1。
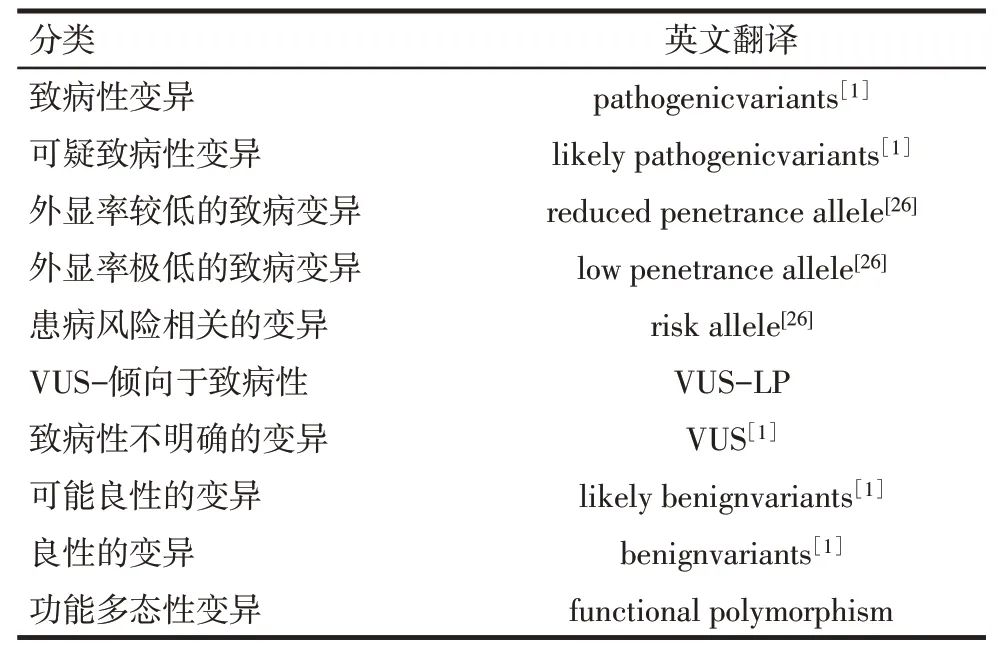
Tab.1 Pathogenicity classification of human gene variants表1 基因變異的致病性的分類
3 意外發現
在進行高通量測序,尤其是全外顯子組測序和全基因組測序時,檢測的范圍不僅覆蓋了與重點關注的表型相關的基因,也覆蓋了其他大量的遺傳信息。在對高通量測序結果分析時,常常可以意外地發現與重點關注表型無關,但是具有重要的提前診斷意義的基因變異。
ACMG最早在2012年開始了關于意外發現的討論[27],并且后續提出了較為體系化的基本原則以及具體的“最少應該報告的意外發現的致病基因范圍”[28-30]。作為基本原則,在高通量測序數據的分析和報告中,至少應該報告外顯率較高、可引起嚴重損害、提前診斷可以使得患者獲益的致病性的變異[29]。
4 案例的臨床表型與可疑基因相關表型譜之間的相符程度
不論是基因變異的致病性分類,還是對案例的檢測結果進行解釋,實際上都需要對可疑基因相關表型與具體案例的臨床信息(包括患者及家屬的臨床表型、家族史、變異的傳遞方式等)之間的相符程度進行評價。本文建議,將臨床符合度劃分為6個級別。
1級,高度特異性相符。第一類:臨床案例具有某些高度特異的表型與疑似基因相關,僅1~2個基因與該表型相關,如有酶活性檢測結果支持的、凝血因子Ⅸ/Ⅹ的活性減低的、具有高度特異性的代謝組學檢測結果支持的。第二類:患者具有一系列特殊表型的組合,特殊的發育里程碑(某些遺傳病的患者在不同的發育階段表現出不同的特殊表型),僅1~2個基因與該表型相關,如符合Prader Willi綜合征的里程碑及系列特征性表型、符合天使綜合征的里程碑及特征性表型。
2級,具有一定特異性的相符。有部分特征表型,但是有3~5個相關基因,如眼皮膚白化病表型;或是有多個基因,但是其中某個基因占比超過50%的,如常染色體隱性遺傳性非綜合征型耳聾患者檢測出1個GJB2的可疑變異。
3級,臨床相符但是沒有特異性。此類表型具有高度的遺傳異質性,缺乏特異性,如智力障礙、自閉癥、癲癇發作。
4級,臨床部分相符,同時存在沖突但不能排除相關性。臨床表型與疑似基因的相關表型有矛盾之處,但是可能用不完全外顯、不完全顯性、伴隨其他疾病來解釋。
5級,有沖突可排除。有部分表型相似,但是有某些表型完全不符,并且存在根本性的矛盾,不能解釋。如臨床疑似肌營養不良,但是免疫組化結果完全排除了抗肌萎縮蛋白病(DMD)的可能性;臨床疑似糖原累積癥,但是酶學檢測酸性麥芽糖苷酶活性完全正常,可排除GAA致病基因的相關性。
6級,不相符。臨床表型與疑似病因完全沒有關系。
5 臨床意義分級的辦法
綜合基因變異的致病性分類、臨床相符程度之后,可進行臨床意義的分級。
1級,很可能解釋臨床信息,或具有極強臨床指導意義的變異。(1)對于案例的診斷有較為明確指導意義的變異。①顯性遺傳基因,分類為致病性或可疑致病性的變異,雜合子、半合子或純合子(需確認半合子、純合狀態同樣可以致病),外顯率不低,臨床相符度為1~4級。須注意,對于既可能顯性遺傳,又可能隱性遺傳的基因,必須在變異水平確認遺傳方式;純合子患者的表型可能與雜合子個體的表型完全不同;個別顯性遺傳基因的半合子或者純合子狀態可能是不致病的。②隱性遺傳基因,分類為致病性或可疑致病性的變異,復合雜合子、純合子或半合子,外顯率不低,臨床相符度1~4級。③線粒體環DNA,分類為致病性或可疑致病性的變異,臨床相符度1~4級。(2)可以完全解釋特定臨床表型的功能多態性位點。(3)對于患者的治療或預后具有明確指導意義的變異。
2級,可能有助于解釋臨床,或具有潛在臨床指導意義的變異。(1)對于案例的診斷有潛在指導意義的變異。包括:①顯性遺傳基因,分類為VUS,臨床相符度為1級;②顯性遺傳基因,分類為VUS-LP,臨床相符度為1~2級;③顯性遺傳基因,分類為VUSLP,臨床相符度為3~4級(可能在獲取了充分的表型信息之后將臨床相符度上升為1~2級,將致病性分類上升為可疑致病變異);④顯性遺傳基因,分類為VUS-LP,臨床相符度為1級,遺傳自否認表型的父母中某一方,表明該變異存在不完全外顯;⑤隱性遺傳基因,純合子,分類為VUS-LP,臨床相符度為1~2級;⑥隱性遺傳基因,復合雜合子,其中1個分類為致病性或可疑致病性,另一個分類為VUS,臨床相符度1~3級;⑦隱性遺傳基因,推測為復合雜合子但未經證實,其中1個分類為致病性或可疑致病性,另一個分類為VUS,臨床相符度1~3級;⑧遺傳方式相符,致病性分類為致病性或可疑致病性,但外顯率較低,臨床相符度1~4級;⑨遺傳方式相符,致病性分類為致病性或可疑致病性,但外顯率極低,臨床相符度1~3級;⑩患病風險相關的變異,已有明確的功能研究的證據,臨床相符度1~3級;?疑似的功能多態性變異,臨床高度相關;?線粒體環DNA,分類為致病性或可疑致病性的變異,外顯率較低,臨床相符度3~4級。(2)可能解釋特定臨床表型的功能多態性位點。(3)對于案例的治療或預后有潛在指導意義的變異。
3級,臨床指導意義不肯定,且不能排除臨床相關性的變異。(1)顯性遺傳基因的VUS-LP,臨床高度相符,遺傳自否認表型的一方父母,沒有證據表明該基因存在不完全外顯,或者沒有證據表明外顯率為100%。(2)隱性遺傳基因,復合雜合子,分類均為VUS,臨床相符度1~4級。(3)隱性遺傳基因,純合子,分類為VUS,臨床相符度1~4級。(4)隱性遺傳基因,推測為復合雜合子但未經證實,分類為VUS,臨床相符度1~4級。(5)隱性遺傳基因的一個雜合致病性/可疑致病性變異,臨床相符。(6)致病性不明確、臨床相關性亦不明確的拷貝數變異(Copy number variation,CNV)。
4級,意外發現的具有潛在臨床干預指導意義的變異。(1)符合ACMG意外發現指南的變異。(2)本實驗室自行定義的具有潛在臨床干預指導意義的變異。
5級,與臨床信息無關的變異,或者分類為良性或可能良性的基因變異。
6 臨床意義分級的應用
本文所提出的臨床意義分級,首先需要厘清基因變異水平的致病性評估與病例的臨床意義的評估(圖1)。本文建議,在基因檢測報告中,不僅描述基因變異的致病性分類,而且應描述基因變異的臨床意義分級,從而提高檢測報告的可理解性。以下通過3個實際案例來闡述如何在臨床檢測中對基因變異進行臨床意義的分級。
(1)案例1。女,4歲。因運動后出現行走姿式異常入院。母親患有“多巴反應性肌張力障礙”,目前服用小劑量左旋多巴,可有效控制肌張力障礙發作。采集患者外周血進行全外顯子組測序,檢測到1個雜合變異:GCH1(NM_000161.2)Exon3:c.468T>A p.Tyr156*;后續檢測患兒母親的外周血,同樣檢測到該變異。
依據ACMG規范進行致病性證據描述與分類如下:該變異為無義變異(編碼蛋白質的第156位氨基酸Tyr變為終止密碼子,預計可使蛋白質翻譯提前終止);HGMD、ESP6500、千人基因組和dbSNP147數據庫均未見收錄;患兒及母親的臨床表型均與GCH1基因相關表型相符。綜合考慮,該變異為致病性變異(按照ACMG的相關證據縮寫為:PVS1、PM2_supporting、PP1)。
依據本建議進行案例水平臨床分級如下:GCH1基因的雜合性失功能性變異已經被反復報道可以引起多巴反應性肌張力障礙伴或不伴高苯丙氨酸血癥(MIM128230),這可以解釋患兒及其母親的臨床表現。綜合考慮,臨床分組為“1級,很可能解釋臨床情況,或具有極強臨床指導意義的變異”。
(2)案例2。男,32歲。主因言語欠清1 d入院。近3個月腦出血2次,發現高血壓2年,痛風5年,目前用4種降壓藥血壓仍控制不佳。采集患者外周血進行全外顯子組測序,檢測到2個雜合變異:ABCG2(NM_004827.2)Exon5:c.421C>A;p.Gln141Lys;KCNH2(NM_000238.3)Exon12:c.2892delC;p.Gly965Glufs*9。
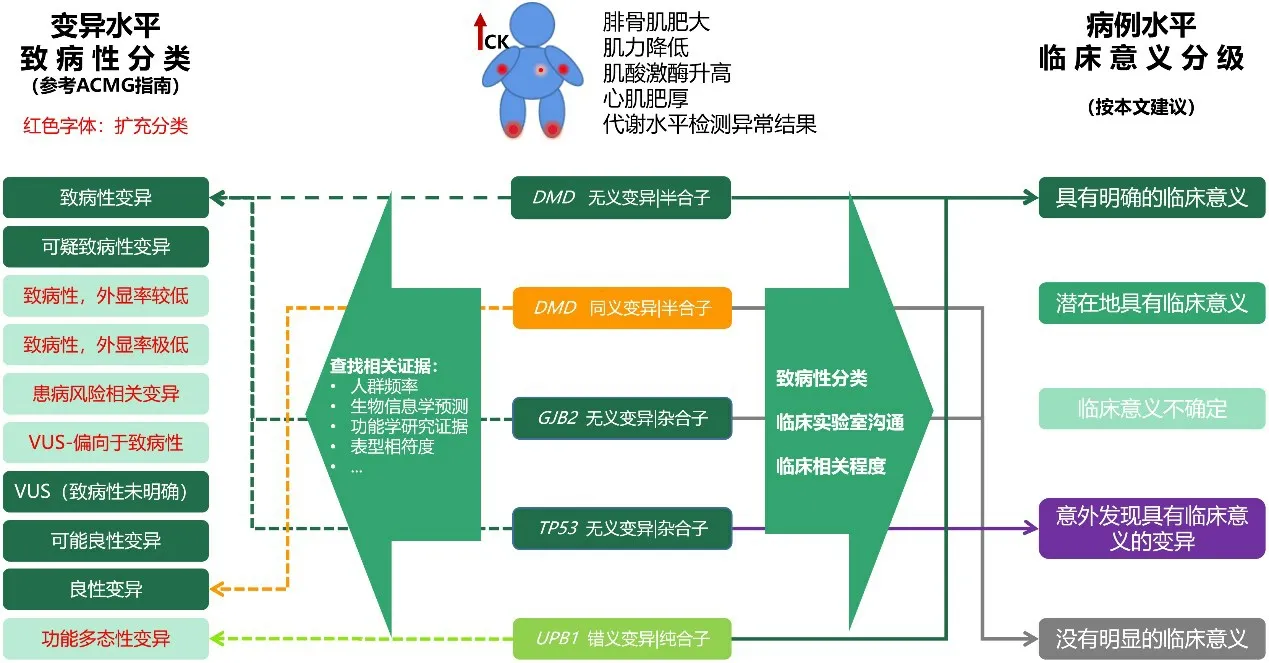
Fig.1 Variant level pathogenicity classification and case level clinical significance grading圖1 基因變異水平的致病性分類與病例水平的臨床意義分級
依據ACMG規范進行致病性證據描述與分類如下:(1)ABCG2基因變異(Gln141Lys)。大量文獻支持該變異與痛風的風險相關,雜合子變異的個體患痛風風險提高1.74倍[31-33]。綜合考慮,該變異分類為患病風險相關的變異。(2)KCNH2基因變異(Gly965Glufs*9)。該變異為框移變異,可引起NMD效應(無義介導的mRNA降解),導致基因功能異常,類似的變異被大量報道可引起長QT綜合征;G1000、ESP、ExAC_ALL與gnomAD_genome_ALL數據庫均未見收錄。綜合考慮,分類為致病性變異(按照ACMG的相關證據縮寫為:PVS1、PM2_supporting、PP4)。
依據本建議進行案例水平臨床分級如下:(1)ABCG2基因變異(Gln141Lys)。該變異已經被明確可以提高痛風癥的風險。患者痛風5年,其他臨床表現可能由痛風繼發(臨床相符但是沒有特異性)。綜合考慮,臨床分組為“2級,可能有助于解釋臨床,或具有潛在臨床指導意義的變異”。(2)KCNH2基因變異(Gly965Glufs*9)。該變異有害性明確,攜帶該變異患長QT綜合征的風險較高,而盡早定期進行心電圖等電生理檢查及采取預防手段,極可能會減少或推遲發病,從而防止產生嚴重后果。綜合考慮,臨床分組為“4級,意外發現的具有潛在臨床干預指導意義的變異”。
(3)案例3。女,2歲,棄嬰。反復癲癇發作。采集外周血進行全外顯子組測序,檢測到SCN3A基因2個雜合變異:SCN3A(NM_006922.3)Exon8:c.905A>G p.Asn302Ser和Exon9 c.968G>T p.Ser323Ile。由于父母樣本不可獲得,故不確認2個變異的相位(incis或者in-trans狀態)。
依據ACMG規范進行致病性證據描述與分類如下:(1)SCN3A基因雜合變異(Asn302Ser)。該變異為錯義變異,有文獻報道該變異可影響電壓門控鈉通道的功能[34];dbSNP147、ESP6500siv2_ALL和千人基因組(1000g2015aug_ALL)數據庫均未見收錄;生物信息學軟件預測其有致病可能性。綜合考慮,該變異分類為可疑致病性(按照ACMG的相關證據縮寫為:PS3_moderate、PP4、PM2_supporting、PP3)。(2)SCN3A基因雜合變異(Ser323Ile)。該變異極為罕見,HGMD、dbSNP147、ESP6500和千人基因組數據庫均未見收錄;生物信息學軟件預測其可能影響mRNA的剪接(dbscSNV_ADA預測值是0.962,dbscSNV_RF預測值是0.692),且預測其有致病可能性。綜合考慮,尚不能確定該變異是否有致病性(按照ACMG的相關證據縮寫為:PM2_supporting、PP3)。
依據本建議進行案例水平臨床分級如下:(1)SCN3A基因雜合變異(Asn302Ser)。SCN3A基因與家族局灶性癲癇伴可變病灶4型(MIM617935)及嬰兒早期癲癇性腦病62型(MIM617938)相關,均為常染色體顯性遺傳,相關表型有發育不良、喂養困難、局灶性癲癇、肌張力降低等,與該臨床信息比較來看,臨床相符但是沒有特異性,該基因變異顯性遺傳且外顯率不低。綜合考慮,臨床分組為“1級,很可能解釋臨床情況,或具有極強臨床指導意義的變異”。(2)SCN3A基因雜合變異(Ser323Ile)。與前一個變異相比,其是否具有致病性尚不能確定,不排除該變異以in-cis或者以in-trans的狀態與前一個變異存在協同作用的可能性。綜合考慮,臨床分組為“2級,可能有助于解釋臨床,或具有潛在臨床指導意義的變異”。
7 其他
目前,絕大多數遺傳病尚沒有或者很少有特異性的治療手段,但對遺傳性腫瘤已經有一些基因變異可以指導此類患者用藥。關于這部分變異的分類,也已在相應的ACMG指南中討論。考慮到這類情況的存在,本文建議在遺傳病的基因變異分類中,也采用相同的原則對具有治療指導意義的變異進行分級,這對于遺傳性腫瘤患兒而言是有價值的。由于對非腫瘤遺傳病有治療指導意義的位點為數不多,本文尚未對這些變異進行展開論述。在臨床基因檢測中如果遇到可指導治療的基因變異,建議參照腫瘤變異分級指南[35-36]。
目前較為主流的臨床全外顯子組測序技術并未特別針對具有治療指導意義的變異進行方法優化,這可能使得部分具有明確用藥指導意義的變異位點不在覆蓋范圍內。全基因組測序技術已經在臨床遺傳基因測序實驗室中有少量的應用[37],主要是針對比較嚴重的疑似遺傳病患者進行檢測,如多發畸形[38]和心臟疾病[39]等多種遺傳病。在全基因組測序結果中就包含了較多具有治療指導價值的遺傳標記[40]。針對全基因組測序中覆蓋的具有治療指導意義的變異位點的臨床報告制定相應的指南,也有助于推動全基因組測序技術的臨床應用。
如同基因往往具有多效性,基因變異也同樣可以具有多效性。例如,GBA基因的致病變異不僅可能與戈謝病(葡糖腦苷脂病)相關,其雜合子還可能提高患早發性帕金森病的風險。因此,在具體臨床案例的分析中,從不同的角度看,同一個基因變異的不同效應可能對應不同的臨床分級。這種情況增加了基因變異臨床分級的復雜度。
由于遺傳病表型的多樣性及醫生對此類疾病認識的相對不足,臨床表型采集難免有不足之處,這些都會對臨床工作中進行基因變異的臨床意義分級造成影響。本方案在臨床實踐過程中必然會存在一些因素使得分級存在一定的主觀性,但是這并不影響本方案能夠使讀者更好地理解遺傳檢測報告。同時,本方案的實施也有助于提高廣大臨床工作者對臨床表型采集的重視程度,促進臨床-實驗室間的溝通,不斷提高臨床意義分級的合理性,從而形成一個良性循環,最終有利于基因檢測報告的解讀及遺傳咨詢。
此外,基因檢測機構要標準化、規范化,要有嚴格的質控標準,并且要不斷更新擴大基因譜檢測范圍,從而發現更多致病性基因,為臨床提供幫助。臨床醫師也要熟悉和掌握基因檢測相關知識,提高解讀基因報告的能力與水平。
執筆:
蔡春泉 天津市兒童醫院(天津大學兒童醫院),天津市兒科研究所,天津市兒童出生缺陷防治重點實驗室
喻長順 天津金域醫學檢驗實驗室,廣州醫科大學金域檢驗學院
舒劍波 天津市兒童醫院(天津大學兒童醫院),天津市兒科研究所,天津市兒童出生缺陷防治重點實驗室
李 光 天津醫科大學基礎醫學院遺傳學系
參與本建議制定的單位及人員(按單位和姓氏拼音排序):
天津大學兒童醫院(陳悅,劉楠,呂玲,王萍,徐曉薇,張玉琴)
天津金域醫學檢驗實驗室(韓曉雪,劉洪洲,劉晴晴)
天津市安定醫院(李潔)
天津市第二人民醫院(李穎)
天津市第三中心醫院(李濤,王鳳梅)
天津市第五中心醫院(劉曉智,姚玲)
天津市第一中心醫院(劉麗,徐鳳琴,張美姿,趙明峰)
天津市婦女兒童保健中心(馮樹人,劉霞,辛力)
天津市海河醫院(陳懷永,吳琦)
天津市環湖醫院(安中平,程秀麗,李慶國,閆華,岳偉)
天津市人民醫院(董昭櫻,王輝,張詩武)
天津市天津醫院(張春智)
天津市胸科醫院(陳慶良,付博,高靜,姜楠,孫大強,徐美林,張穎)
天津市眼科醫院(李津,李軒,史學鋒)
天津市中西醫結合醫院(崔云峰,王玉水)
天津市中心婦產科醫院(常穎,羅海寧,任晨春,田秀英,張玥)
天津市中醫藥研究院附屬醫院(劉鵬飛)
天津醫科大學第二醫院(劉長山,王建梅,王雪艷)
天津醫科大學基礎醫學院遺傳學系(李衛東,時文濤,王峰)
天津醫科大學眼科醫院(魏瑞華)
天津醫科大學腫瘤醫院(任麗,于津浦,趙海豐)
天津醫科大學總醫院(姜麗紅,李增彥,史云芳,張穎,鄭榮秀)
天津中醫藥大學第一附屬醫院(夏天)
猜你喜歡
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
數學小靈通(1-2年級)(2021年4期)2021-06-09 06:25:56
中學生數理化·七年級數學人教版(2019年4期)2019-05-20 10:06:32
中學生數理化·七年級數學人教版(2018年6期)2018-06-26 08:36:06
初中生世界·七年級(2017年9期)2017-10-13 22:27:46
財經(2017年2期)2017-03-10 14:35:35
財經(2016年15期)2016-06-03 07:38:02
海峽科技與產業(2016年3期)2016-05-17 04:32:12
