Wiskott-Aldrich綜合征的臨床特征與基因表型分析
2021-05-20 03:46:42金玉婷顧靜文馬典慶劉野天陳天平
安徽醫(yī)專學(xué)報(bào) 2021年2期
金玉婷 王 敏 韋 楠 顧靜文 馬典慶 劉野天 陳天平
Wiskott-Aldrich綜合征(WAS)又稱濕疹血小板減少伴免疫缺陷綜合征,為一種罕見的X連鎖(Xp11.22-23)隱性遺傳性疾病,其發(fā)病率約為(1~10)/100萬[1-2]。該病多因感染和血小板減少就診,易與免疫性血小板減少癥混淆,早期診斷可指導(dǎo)患兒行造血干細(xì)胞移植(HSCT)以改善預(yù)后,故對該病進(jìn)行及時(shí)、準(zhǔn)確的診斷顯得非常重要。本文分析我院3例WAS患兒臨床特征及基因檢測結(jié)果,初步探討該病的治療方法,以提高對本病的認(rèn)識(shí)。
1 材料和方法
1.1 研究對象 先證者1,男,2月6天,初診時(shí)因皮膚出血點(diǎn)、肛周皮膚紅腫入院,入院后行骨穿等相關(guān)檢查,擬診為免疫性血小板減少癥,后因療效欠佳,完善第二代基因測序(NGS),確診為WAS,予轉(zhuǎn)外院行HSCT。先證者2,男,1月7天,初診時(shí)即有大便帶血、皮膚出血點(diǎn)等嚴(yán)重出血傾向,在我科完善相關(guān)檢查后擬診免疫性血小板減少癥,后因療效欠佳完善NGS檢查,確診為WAS,于等待HSCT期間合并嚴(yán)重感染死亡。先證者3,男1月6天,因腹瀉、皮膚潰瘍及皮膚出血點(diǎn)就診,完善骨穿及NGS檢查,確診為WAS,隨后聯(lián)系外院行HSCT。
1.2 一般實(shí)驗(yàn)室檢查 3例患兒入院后均完成了一般實(shí)驗(yàn)室檢查,包括:血常規(guī)、免疫球蛋白定量、炎癥反應(yīng)指標(biāo)、EBV-DNA及骨髓穿刺檢查等,相關(guān)檢查方法均為臨床實(shí)驗(yàn)室常用檢查,在此不再一一贅敘,相關(guān)檢查結(jié)果見表1。
1.3 第二代基因測序 3例患兒均使用NGS的方法進(jìn)行了全外顯子掃描(WES)。具體分為以下三個(gè)步驟:突變篩查、基因數(shù)據(jù)分析和疑似致病突變驗(yàn)證。①突變篩查:采用IDT The x Gen Exome Research Panel v1.0全外顯子捕獲芯片,經(jīng)Illumina Nova Seq 6000系列測序儀測序,目標(biāo)序列測序覆蓋度≥99%。②基因數(shù)據(jù)分析:使用智因東方“全譜遺傳病精準(zhǔn)診斷云平臺(tái)系統(tǒng)”分析篩選,結(jié)合致病突變數(shù)據(jù)庫、正常人基因組數(shù)據(jù)庫、已知四千種遺傳病臨床特征數(shù)據(jù)庫等基因數(shù)據(jù)分析算法,對發(fā)現(xiàn)的基因變異進(jìn)行分級(jí),變異分級(jí)采用ACMG(美國醫(yī)學(xué)遺傳學(xué)會(huì))基因變異分級(jí)體系。③疑似致病突變驗(yàn)證:對目標(biāo)序列進(jìn)行PCR擴(kuò)增,使用ABI3730測序儀進(jìn)行Sanger測序驗(yàn)證,并經(jīng)基因序列分析軟件獲得基因驗(yàn)證結(jié)果。
2 結(jié) 果
2.1 3 例先證者臨床表現(xiàn)及一般實(shí)驗(yàn)室檢查結(jié)果 3例患兒初診時(shí)均有濕疹表現(xiàn),3例初診時(shí)均有出血表現(xiàn),先證者1和先證者3出血部位在皮膚黏膜,先證者2有皮膚黏膜和消化道出血。先證者1既往有肛周感染、敗血癥病史,先證者2和先證者3既往有肺炎感染病史。3例患兒初診時(shí)擬診為免疫性血小板減少癥,經(jīng)糖皮質(zhì)激素、丙球治療,血小板計(jì)數(shù)均不能恢復(fù)。3例患兒家系中均無血小板減少及其他血液系統(tǒng)疾病家族史,亦無近親結(jié)婚史。3例先證者的一般實(shí)驗(yàn)室檢查結(jié)果,見表1。
2.2 基因檢測及家系驗(yàn)證結(jié)果 先證者1的WAS突變基因?yàn)榈?090位核苷酸C>T(p.R364X)[c.1090C>T, p.R364X],造成編碼蛋白質(zhì)的第364位氨基酸由精氨酸(Arg,R) 變?yōu)榻K止子(X,Ter)(圖1A),該突變?yōu)榘牒献油蛔儯瑏碓从谀赣H,見圖2A。先證者2的WAS突變基因突變?yōu)榈?號(hào)外顯子411號(hào)核苷酸和4222號(hào)核苷酸之間插入序列(CGGGCCCT)[c.411(exon4)_c.412(exon4)ins CGGGCCCT](圖1B),突變類型為半合子突變,來源于母親(圖2B)。先證者3的WAS基因突變位為第7號(hào)外顯子631位核苷酸C>T [c.631C>T(exon 7),pp.R211X, 292](圖1C),突變類型為半合子突變,家系驗(yàn)證結(jié)果提示該突變?yōu)樾律蛔儯▓D2C)。

表1 一般實(shí)驗(yàn)室檢查結(jié)果
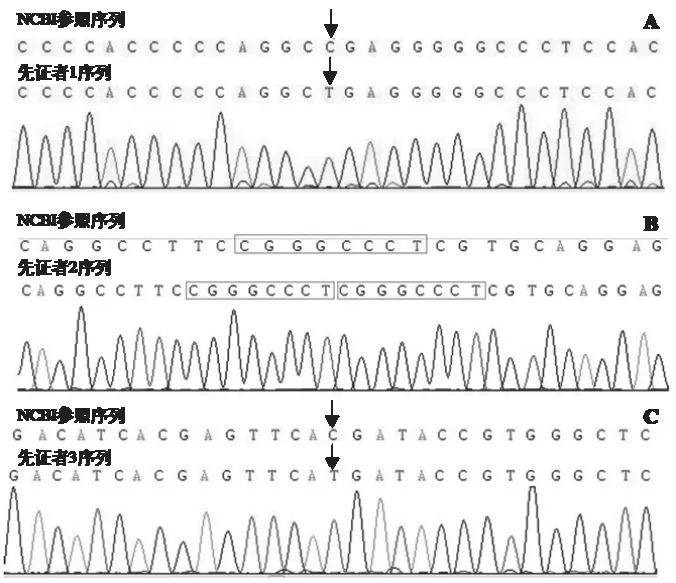
圖1 WAS基因檢測峰圖
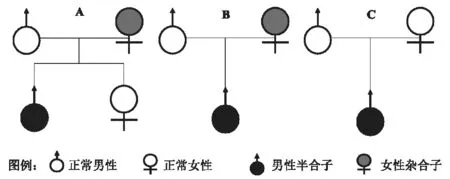
圖2 WAS基因表型家系圖
3 討 論
Wiskott-Aldrich綜合征(WAS)為一組X-連鎖隱性遺傳性疾病,以濕疹、小血小板性血小板減少和反復(fù)感染為主要臨床表現(xiàn),男性發(fā)病為主,發(fā)病率約為1/100萬~10/100萬,臨床相對少見[3]。該病因出血、反復(fù)感染,導(dǎo)致患兒生長發(fā)育受阻、生存質(zhì)量顯著降低,據(jù)統(tǒng)計(jì),未經(jīng)治療的WAS患兒,其中位生存年齡僅15歲[4]。雖然該病為單基因遺傳病,但因其突變類型多變,因此其臨床表現(xiàn)和疾病嚴(yán)重程度存在很大的異質(zhì)性。雖然當(dāng)前可以通過基因檢測確診WAS,但因其發(fā)病率非常低,而其他一般性實(shí)驗(yàn)室檢查僅表現(xiàn)為血小板減少,故易誤診為免疫性血小板減少癥(ITP)[3]。本文所述3例WAS患者,初診時(shí)均擬診為ITP,后因治療效果欠佳方經(jīng)基因檢測確診為WAS。
WAS致病基因位于X染色體p11.22-23,含12個(gè)外顯子,長約9 kb,編碼由502個(gè)氨基酸組成的胞漿蛋白,稱WAS蛋白(WASP)[5]。WAS基因突變引起WASP結(jié)構(gòu)和/或功能異常,導(dǎo)致免疫細(xì)胞的遷移、免疫突觸形成及細(xì)胞骨架重塑等結(jié)構(gòu)和功能障礙,影響機(jī)體的特異性及非特異性免疫功能[2],故WAS患兒一般有反復(fù)感染病史。血小板減少、免疫缺陷和濕疹是WAS經(jīng)典的臨床表現(xiàn)三聯(lián)癥,但在新生兒期,這些臨床特征可不典型。本文所述3例患兒,初診時(shí)均年齡較小,3例患兒在就診時(shí)均有血小板減少、濕疹和既往感染病史,符合WAS疾病的一般臨床表現(xiàn)。此外,WAS的血小板減少常為小血小板體積性血小板減少,但本文所述3例患兒,僅1例表現(xiàn)為血小板體積減小,其余2例血小板體積均正常;我們既往報(bào)道1例WAS合并石骨癥的血小板減少癥患兒,其血小板體積亦正常[6],這些提示臨床上不能因血小板體積正常而輕易排除WAS的診斷。
WAS的治療方案因其疾病的異質(zhì)性而又所不同,造血干細(xì)胞移植(HSCT)是目前根治WAS的主要手段[2]。對于血小板減少癥的患兒,糖皮質(zhì)激素及丙球沖擊治療效果均不理想,血小板輸注一般僅推薦用于發(fā)生嚴(yán)重大出血的患兒。因WAS患兒的血小板減少與脾臟破壞增多有關(guān),故脾切除可一定程度上增加WAS的血小板數(shù)量并增加長期生存率,但脾切除可能會(huì)增加HSCT后的相關(guān)并發(fā)癥[7-8]。據(jù)統(tǒng)計(jì),未行HSCT的WAS患兒最終將死于感染(44%)、出血(23%)和惡性腫瘤(26%),而HSCT治療后的WAS患兒長期生存率可達(dá)80%以上。影響HSCT治療效果的關(guān)鍵因素在于移植的時(shí)間,目前認(rèn)為5歲以前HSCT的治療效果要優(yōu)于5歲以后,且5歲前無關(guān)供者HSCT的療效與同胞相合供者的療效相差不大。因此,對于WAS患兒的早期診斷顯得尤為重要。本文所述3例患兒初診時(shí)曾行激素及丙球沖擊治療,但療效均不佳,這與WAS的疾病特點(diǎn)有關(guān)。其后2例患兒行HSCT,1例于等待移植過程中死亡。截止到目前為止,2例HSCT后患兒反復(fù)感染和濕疹的臨床表現(xiàn)基本消失,血小板計(jì)數(shù)基本恢復(fù)正常。
綜上所述,本研究報(bào)道了3個(gè)經(jīng)NGS基因確診的WAS病例,分析并探討了WAS的臨床特征。對于男性嬰幼兒、尤其是小嬰兒,如出現(xiàn)血小板減少合并反復(fù)感染、濕疹,無論血小板體積是否減少,均應(yīng)警惕WAS的可能性。基因檢測是目前診斷WAS的金標(biāo)準(zhǔn),HSCT是其確切有效的治療手段。
