蒸氨法制備Ni/SiO2 催化劑及其在2-MF 加氫制2-MTHF反應中的應用
2021-02-06 07:44:44王英文張雅靜王康軍談立美陳姝穎
燃料化學學報 2021年1期
關鍵詞:催化劑
王英文 ,張雅靜 ,王康軍 ,談立美 ,陳姝穎
(沈陽化工大學 化學工程學院,遼寧 沈陽 110142)
2-甲基四氫呋喃(2-MTHF)是一種重要的醫藥中間體和新型綠色溶劑。與傳統溶劑四氫呋喃(THF)相比,2-MTHF 沸點適中、易于與水分離,且具有合適的路易斯堿強度,因而被廣泛的應用于有機化學和商業化的樹脂工業中[1,2]。2-MTHF 燃燒性能優良,還可以用作汽油添加劑。在發動機正常運轉的前提下,2-MTHF 在汽油中的添加體積占比可達到60%[3]。因而,2-MTHF 合成研究受到廣泛關注。
2-MTHF 可由生物質及其衍生物轉化生成。根據原料種類,合成2-甲基四氫呋喃有三種方法,二元醇法、內酯法和糠醛法[4]。Donald 等[5]采用2-甲基-1,4-丁二醇為原料,在五乙氧基磷的催化下,分子內脫水制得2-甲基四氫呋喃,其收率達69.2%。Saneo 等[6]采用內酯為原料,在SiHCl3存在條件下,以γ-rays 或紫外光照射內酯,制備出2-甲基四氫呋喃,收率達82.3%。以糠醛為原料合成2-甲基四氫呋喃可分為一步法和兩步法:一步法是將糠醛催化加氫一步得到2-MTHF。Fang 等[7]以糠醛為原料,將雙固體催化劑Cu/SiO2和Pd/SiO2填充于一個反應器的兩段中,一步加氫合成2-MTHF,常壓下其產率高達97.1%。二步法分為兩種路線,一種流程是糠醛先通過Cannizzaro 反應生成糠醇,再將糠醇催化加氫生成2-甲基四氫呋喃[8]。Proskuryakov 等[9]采用Raney-Ni 催化劑使得糠醇轉化為2-MTHF,2-MTHF 的產率為38.5%。另一種路線是糠醛先催化加氫得到2-甲基呋喃(2-MF),然后2-MF 催化加氫得到2-MTHF。Ni 基催化劑對于2-MF 呋喃環上C=C 加氫具有良好的催化作用,催化性能受到制備方法和反應條件的影響。李增杰等[10]采用浸漬法制備Ni/Al2O3催化劑,用于2-MF 液相加氫制2-MTHF 反應。在壓力為3 MPa,反應溫度為150 ℃,攪拌速率為1000 r/min 的條件下,2-MTHF的收率可達96%以上。Ding 等[11]采用溶膠-凝膠法制備Ni/SiO2催化劑用于2-MF 氣相加氫制2-MTHF 反應,在反應溫度為180 ℃,反應壓力為1.6 MPa 下,Ni 負載量為25%時,2-MTHF 的產率可達85%;同時研究結果表明,催化劑的穩定性較差,在反應10 h 后,催化劑活性迅速下降。近來文獻報道采用蒸氨法制備Ni/SiO2催化劑可以獲得較小的晶粒尺寸、較強的金屬-載體相互作用,在不同的反應中表現出高活性和高穩定性的特點。Ashok 等[12]采用蒸氨法和浸漬法分別制備Ni/SiO2催化劑,均應用于生物炭重整反應,蒸氨法制備的催化劑形成層狀硅酸鎳結構,展現較強的金屬-載體相互作用,因而展現出較好的催化性能和熱穩定性。Zhang 等[13]采用蒸氨法制備了Ni/mSiO2催化劑用于苯甲酸加氫反應,發現該催化劑Ni 晶粒高度分散、晶粒尺寸小,且金屬與載體之間存在較強的相互作用,展現了優異的催化性能和穩定性。
本研究以硝酸鎳為鎳源,硅溶膠為硅源,采用蒸氨法制備Ni/SiO2催化劑,應用于2-MF 加氫合成2-MTHF 反應。考察了焙燒溫度和反應條件對催化劑性能的影響,并考察了催化劑穩定性,探討了催化劑失活原因。
1 實驗部分
1.1 催化劑的制備
蒸氨法制備Ni/SiO2催化劑過程如下:稱取一定質量的Ni(NO3)2·6H2O 中加入去離子水配成一定濃度的硝酸鎳溶液,放在水浴鍋中室溫攪拌至完全溶解。在溶解后的硝酸鎳溶液中加入定量的濃氨水,室溫條件下密封攪拌10 min,將定量的堿性硅溶膠滴加到鎳氨溶液中,在室溫條件下密封攪拌4 h。將混合物在80 ℃條件下加熱,待混合物pH 值降為6.0-7.0 時,停止加熱。經去離子水多次洗滌抽濾后,120 ℃干燥12 h,分別在400、500、600 和700 ℃條件下焙燒4 h,得到催化劑。催化劑的金屬負載量為30%(按照NiO 質量分數計)。
1.2 催化劑的表征
BET 測試采用Autosorb-iQ-C 型全自動物理化學吸附儀進行催化劑的比表面積和孔結構測定。稱取0.05 g 樣品在300 ℃真空脫氣處理3 h,在液氮溫度下吸附,測得催化劑的比表面積、孔容和孔結構。
XRD 分析采用Bruker 公司D8 Advance 型X 射線衍射儀,Cu 靶Kα 線,管電壓為50 kV,管電流為300 mA,10°-90°掃描,掃描速率10 (°) /min。
H2-TPR 通過美國康塔公司ChemBET Pulsar TPR/TPD 自動化學吸附儀測試,稱取0.05 g 樣品裝入U 型石英玻璃管中,在400 ℃的He 氣氛預處理30 min,溫度降至50 ℃,切換成體積比v(H2)∶v(N2) = 1∶9 的氫氮標準混合氣,氣流量均為30 mL/min,10 ℃/min 的速率升溫至900 ℃,記錄樣品的H2-TPR 譜圖。
NH3-TPD 通過美國康塔公司ChemBET Pulsar TPR/TPD 自動化學吸附儀測試,稱取200 mg 催化劑樣品裝入石英玻璃管中,在550 ℃條件下通入He 吹掃30 min,將至50 ℃,切換NH3吸附60 min,之后改為He 在50 ℃吹掃樣品1 h,最后以10 ℃/min升溫至550 ℃,記錄NH3脫附曲線和溫度曲線。
采用X 射線光電子能譜(XPS, ThermoES CaLAB 250 Al 靶)表征催化劑的表面化學狀態及表面原子比。
催化劑樣品的熱失重分析在Pekin Elmer 公司的Pvris Diamond 型TG 熱分析儀器進行,空氣流量100 mL/min,以10 ℃/min 升溫速度到1000 ℃。
1.3 催化劑的活性評價
采用固定床反應器對催化劑性能進行評價。將焙燒后催化劑壓片(10 MPa 壓力)、過篩,篩取粒徑20-40 目顆粒進行活性評價。將1 g 的催化劑裝入反應管中間部位,兩端以石英砂填料固定。反應前通入氫氮混合氣(N2∶H2= 8∶2),由20 ℃以10 ℃/min 升溫至600 ℃常壓還原4 h。催化劑還原后,在一定溫度、壓力條件下,保持恒溫恒壓進行催化加氫反應。采用氣相色譜SP-3420A 分析反應后產物的組成。其中色譜條件為:色譜柱:KB-1,柱溫為50-230 ℃,程序升溫;進樣器溫度為250 ℃;FID 檢測器為250 ℃。色譜柱的初始溫度為50 ℃,在初始溫度下維持2 min,由50 ℃以10 ℃/min 升溫至230 ℃,并在230 ℃下維持2 min。采用面積歸一化法進行定量分析。
2 結果與討論
2.1 Ni/SiO2 催化劑的比表面積及孔結構
不同焙燒溫度條件下制備的Ni/SiO2催化劑N2吸附-脫附等溫線和孔分布曲線見圖1,比表面積、孔徑和孔容見表1。

圖 1 不同焙燒溫度制備的Ni/SiO2 催化劑的N2 吸附-脫附等溫線(a)和BJH 孔徑分布(b)Figure 1 N2 adsorption-desorption isotherms (a) and BJH pore size distributions (b) of Ni/SiO2 catalysts
由圖1(a)可知,每條等溫線均屬于Ⅳ型,表明催化劑具有介孔結構[14]。在p/p0大于0.5 時,催化劑的吸附-脫附曲線均出現了一個遲滯環,根據IUPAC 規定可知,這些遲滯環均屬于H2 型,表明孔的結構是口小腔大的“墨水瓶”狀的孔[15]。由圖1(b)可知,催化劑的孔徑都分布在2-8 nm,為雙孔結構。隨著焙燒溫度升高,孔徑逐漸變大。這種變化趨勢是合理的,焙燒溫度升高會將小孔之間的孔壁燒塌陷,從而孔徑增大。孔徑的具體數值見表1,700 ℃焙燒制備的Ni/SiO2催化劑孔徑最大,為5 nm。制備催化劑的焙燒溫度對催化劑的比表面積影響較大,隨著焙燒溫度的升高,比表面積明顯下降。在500 ℃或600 ℃下焙燒時,催化劑的比表面積比較接近。
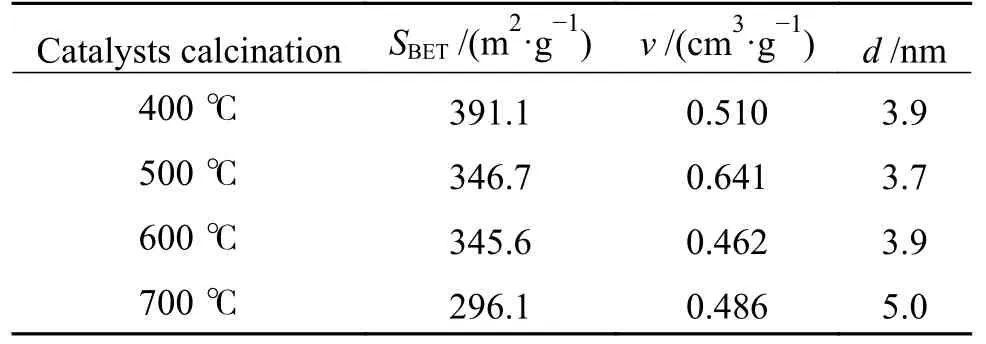
表 1 不同焙燒溫度制備的Ni/SiO2 催化劑的比表面積、孔容和孔徑Table 1 Specific surface area, pore volume and pore sizes of different catalysts
2.2 Ni/SiO2 催化劑的XRD 表征
圖2 為不同焙燒溫度制備的Ni/SiO2催化劑的XRD 譜圖。由圖2(a)可知,焙燒后各個催化劑均在23°出現較寬的衍射峰,表明存在無定形的同時,催化劑均在37.3°、43.7°、62.9°和72.5°時出現Ni3Si4O12H2的特征峰(PDF#43-0664)[17],表明樣品中存在層狀硅酸鎳,這與文獻結果一致[18]。蒸氨法制備的Ni/SiO2催化劑,主要以層狀硅酸鎳的形式存在,這種層狀結構是在催化劑沉淀過程中形成的。焙燒后催化劑的XRD 譜圖中沒有觀察到明顯的NiO 的衍射峰,這可能是NiO 的含量較少或者高度分散所致。由圖2(b)可知,還原后的催化劑在2θ = 23°時出現無定形SiO2的衍射峰,在37.3°和62.9°仍然存在層狀硅酸鎳的特征峰,表明催化劑在還原后保持了層狀硅酸鎳的結構。同時,XRD 譜圖中44.5°、51.8°和76.4°時出現較寬且弱的Ni 的特征峰[19],表明還原后鎳晶粒尺寸較小,高度分散于SiO2載體上。隨著焙燒溫度升高,Ni 的衍射峰變強變窄,表明Ni 的晶粒尺寸逐漸增大。

圖 2 不同焙燒溫度制備的Ni/SiO2 催化劑的XRD 譜圖Figure 2 XRD patterns of Ni/SiO2 catalysts prepared at different calcination temperatures
2.3 Ni/SiO2 催化劑的H2-TPR
圖3 為不同焙燒溫度制備的Ni/SiO2催化劑H2-TPR 譜圖。由圖3 可知,在溫度為400-870 ℃,每個H2-TPR 曲線中均存在較寬的還原峰。通過高斯分峰,還原峰可以分為兩個。在400-500 ℃的還原峰,歸因于與SiO2有相互作用的束縛態Ni2+的還原;溫度在500-870 ℃的還原峰,可以歸因于位于層狀硅酸鎳中的Ni2+的還原[20,21]。隨著焙燒溫度升高,高溫還原峰的還原溫度呈現升高的趨勢,這說明提高焙燒溫度,可以加強催化劑金屬-載體之間相互作用。另外,和本實驗室采用溶膠凝膠方法制備的Ni/SiO2催化劑還原性能相比,還原溫度明顯升高[11],這表明蒸氨法制備的催化劑具有較強的金屬載體相互作用[22]。溶膠凝膠法制備的該催化劑焙燒后,XRD 譜圖中顯示的是NiO 和SiO2的衍射峰,而蒸氨法制備的催化劑焙燒后是層狀硅酸鹽結構,而這種相互作用有利于束縛Ni 顆粒,從而阻止催化劑快速燒結,這可能是不同焙燒溫度的催化劑仍保有較高的催化活性的原因。與此同時,也是因為形成這種結構,鎳的晶粒尺寸小且分散性好。
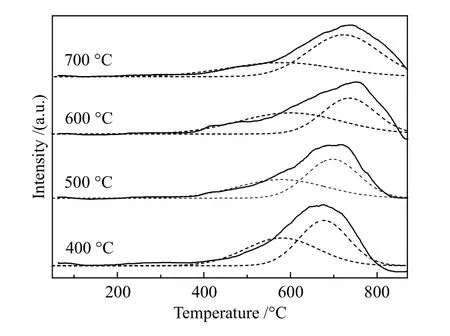
圖 3 不同焙燒溫度制備的Ni/SiO2 催化劑的H2-TPR 譜圖Figure 3 H2-TPR profiles of Ni/SiO2 catalysts prepared at different calcination temperatures
2.4 Ni/SiO2 催化劑的NH3-TPD
催化劑表面酸性由NH3-TPD 表征。不同焙燒溫度的Ni/SiO2催化劑的NH3-TPD 譜圖見圖4,每個催化劑均在100-200 ℃存在一個較寬的NH3脫附峰,表明催化劑表面均存在弱酸中心。隨著焙燒溫度的升高,脫附峰的位置不變,表明弱酸中心強度基本不變,但催化劑的NH3脫附峰的峰面積呈現減小的趨勢,說明催化劑的表面酸量隨著焙燒溫度的升高而呈現減小的趨勢。催化劑的酸量如表2 所示,當焙燒溫度為500 ℃時,催化劑的酸量達0.411 mmol NH3/gcat。據文獻報道,蒸氨法制備的Ni/SiO2催化劑,其酸性位來源于層狀硅酸鎳表面或邊緣的Ni(Ⅱ)[18]。焙燒溫度對催化劑的結構產生影響,隨著焙燒溫度的升高,催化劑的比表面積減小,從而導致催化劑中層狀硅酸鎳表面的Ni(Ⅱ)減少,最終導致酸量減少。因此,焙燒溫度會影響催化劑表面的酸性位數量。
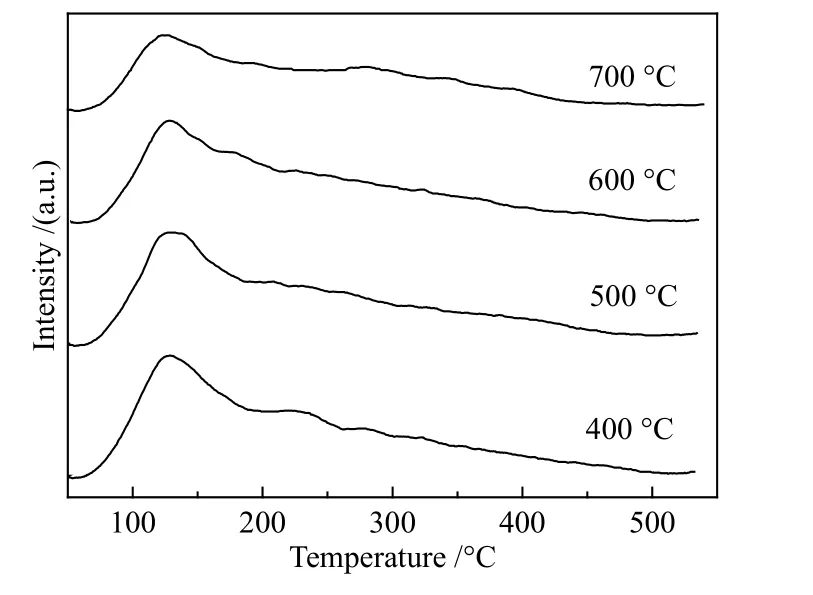
圖 4 不同焙燒溫度制備的Ni/SiO2 催化劑的NH3-TPD 譜圖Figure 4 NH3-TPD profiles of Ni/SiO2 catalysts prepared at different calcination temperatures

表 2 不同焙燒溫度制備的Ni/SiO2 催化劑的酸量Table 2 Total acidity amount of different catalysts
2.5 Ni/SiO2 催化劑的性能
在反應溫度為120 ℃,反應壓力為1.6 MPa,H2/喃(物質的量比)為5.4,空速為1.6 h-1,反應持續運行10 h,評價不同焙燒溫度制備的Ni/SiO2催化劑催化2-甲基呋喃加氫制2-甲基四氫呋喃的催化性能,結果見圖5。由圖5 可知,在不同的焙燒條件下,催化劑均具有較好的活性,2-甲基呋喃均完全轉化,即2-甲基呋喃的轉化率達100%,而2-MTHF 的選擇性則不同。產物除了2-MTHF,還有戊醇、2-戊醇等副產物。當焙燒溫度從400 ℃升高到700 ℃時,2-甲基四氫呋喃的選擇性呈先增加后降低的趨勢,當焙燒溫度為500 ℃時,2-甲基四氫呋喃的選擇性最高,達89.5%。不同焙燒溫度制備的催化劑均保持較高的催化活性,表明催化劑均未過度燒結,因而均保持較好的活性[23]。Dong等[18]認為,蒸氨法制備的催化劑有利于形成層狀硅酸鹽結構,而這種結構可提高金屬與載體之間相互作用,防止金屬顆粒的移動,抑制催化劑的快速燒結。不同焙燒溫度所制備的催化劑對于2-甲基四氫呋喃的選擇性不同,可能與催化劑的表面酸性有關。催化劑表面的酸性位過多會促進2-甲基呋喃中呋喃環上C-O 斷裂,生成副產物,降低2-甲基四氫呋喃的選擇性[24]。600 和700 ℃焙燒后的催化劑,酸量較500 ℃催化劑酸量低,但是選擇性也略低,這表明酸性不是影響選擇性的唯一因素,可能存在金屬酸性協同作用。
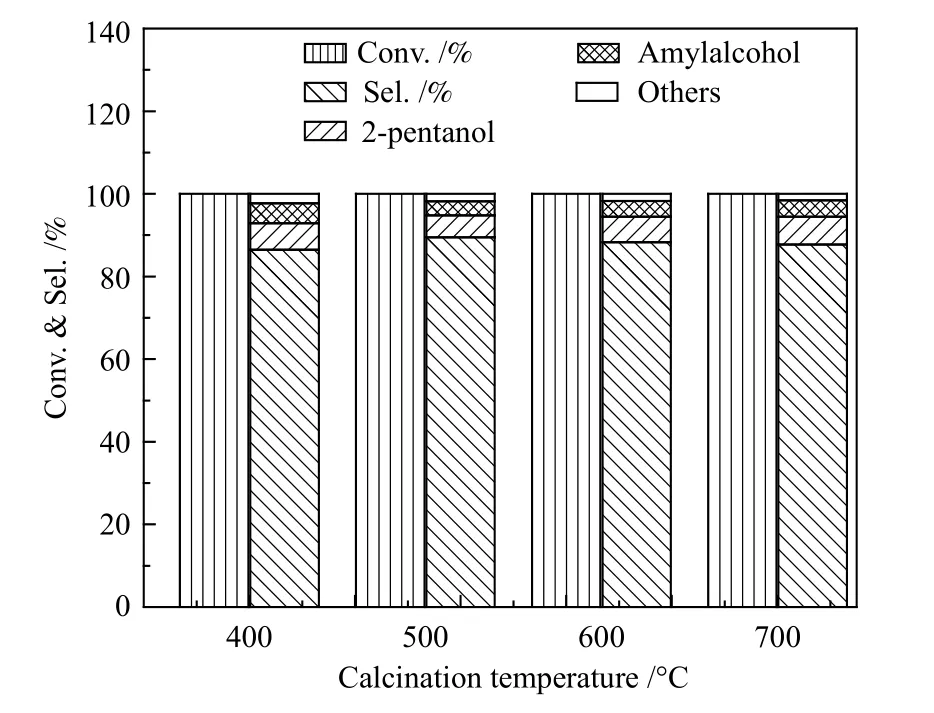
圖 5 不同焙燒溫度制備的Ni/SiO2 催化劑的性能Figure 5 Catalytic performance of Ni/SiO2 catalysts prepared at different calcination temperatures
2.6 反應條件對催化劑性能的影響
2-MF 催化加氫制2-MTHF 是放熱反應,反應過程中受溫度影響較大。表3 為不同反應條件下催化劑的催化性能,由表3 可知,在110-180 ℃,2-甲基呋喃的轉化率均達到100%,2-甲基四氫呋喃的選擇性呈現先增大后減小的趨勢。當反應溫度達到160 ℃時,2-甲基四氫呋喃的選擇性最大,為95.3%。2-MF 催化加氫制2-MTHF 是分子數減小的反應,增大反應壓力對成產物有利,在研究范圍內(1.2-1.6 MPa),反應壓力對Ni/SiO2催化劑性能的影響較小。較高H2/喃物質的量比會提升2-MTHF的選擇性。
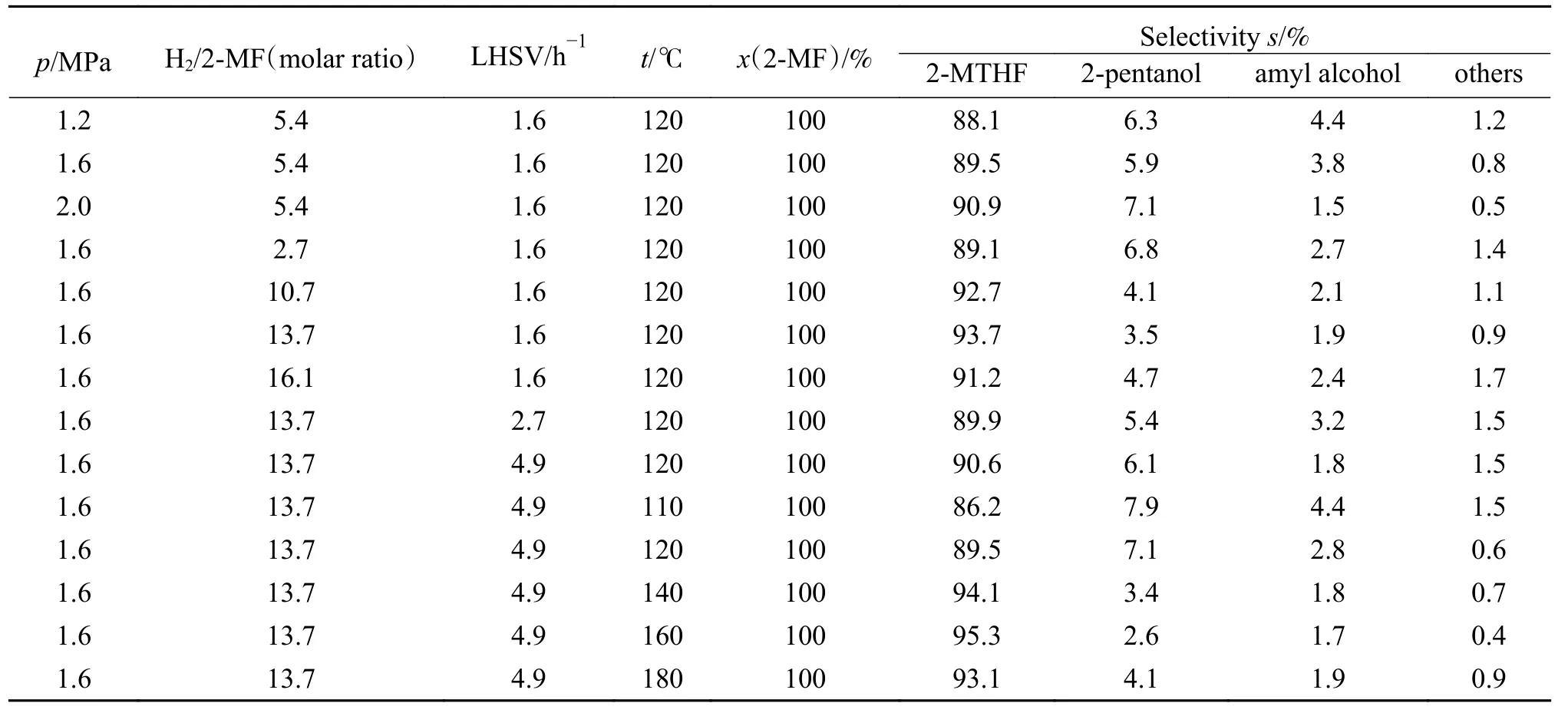
表 3 不同反應條件下催化劑的催化性能Table 3 Catalytic performance of Ni/SiO2 catalyst under different reaction conditions
2.7 催化劑的穩定性能
為了研究催化劑的穩定性,測試500 ℃焙燒條件制備的Ni/SiO2催化劑的30 h 反應性能,結果見圖6。反應溫度為160 ℃,壓力1.6 MPa,空速LHSV 為1.6 h-1,氫氣和2-甲基呋喃的物質的量比為5.4,持續反應15 h,一直維持較高的活性,轉化率大于99%;在15 h后,催化劑活性下降較快。反應30 h,2-甲基呋喃的轉化率下降到75%。在反應時間內,2-甲基四氫呋喃的選擇性一直比較穩定。該結果表明,蒸氨法所制備的催化劑穩定性高于溶膠凝膠法所制備Ni/SiO2催化劑的穩定性[11]。
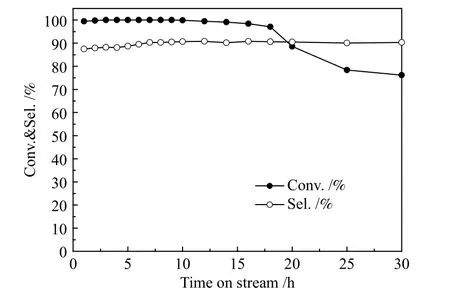
圖 6 Ni/SiO2 催化劑的穩定性Figure 6 Stability of Ni/SiO2 catalyst (performance vs.time on stream)
2.8 Ni/SiO2 催化劑的失活原因
圖7 為Ni/SiO2催化劑的XRD 譜圖,對比圖7還原后與使用后Ni/SiO2催化劑的XRD 譜圖發現,43.7°處Ni 的衍射峰強度明顯增強,表明Ni 晶粒尺寸變大。
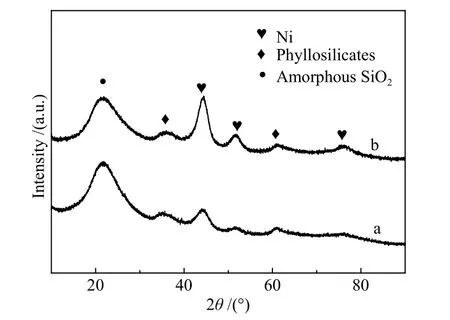
圖 7 Ni/SiO2 催化劑的XRD 譜圖Figure 7 XRD patterns of Ni/SiO2 catalysts
使用后催化劑表面的C 原子的含量(XPS)增加到還原催化劑的3 倍左右,這表明反應后Ni/SiO2催化劑表面被含碳有機物覆蓋或者積炭[25](見表4)。

表 4 Ni/SiO2 催化劑表面原子百分比Table 4 Atom ratio on the surface of Ni/SiO2 catalysts
由圖8 可知,使用后催化劑的TG 曲線從室溫到650 ℃失重15%左右,主要是催化劑表面有機物的脫除所致。這表明反應過程中催化劑表面被部分沉積物種所覆蓋。結合XPS 結果,有機物種的沉積在催化劑表面是催化劑失活的主要原因。
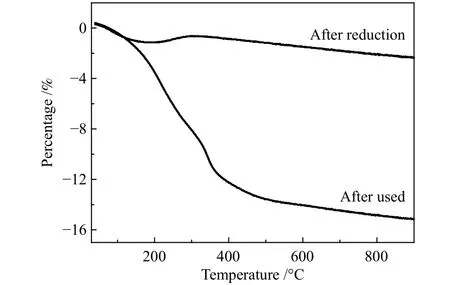
圖 8 Ni/SiO2 催化劑的熱失重曲線Figure 8 TG curves of the Ni/SiO2 catalysts
3 結 論
催化劑的焙燒溫度影響Ni/SiO2催化劑的晶粒尺寸、還原性能、金屬載體相互作用以及催化劑的表面酸性。
隨著焙燒溫度的升高,催化劑晶粒尺寸逐漸增大、還原峰溫度升高、金屬載體相互作用逐漸增強,表面酸性逐漸下降。
催化劑在優化條件下,反應15 h 比較穩定。Ni晶粒長大和有機物覆蓋是催化劑失活的原因。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
