一種檢測生活飲用水和污水中生物有效性銅離子的微生物報告菌株的構建
2020-06-11 11:48:58周懷彬王娟娟趙建國洪旭芬龐一林李江輝譚國強
溫州醫科大學學報 2020年5期
周懷彬,王娟娟,趙建國,洪旭芬,龐一林,李江輝,譚國強
(溫州醫科大學檢驗醫學院生命科學學院檢驗醫學教育部重點實驗室浙江省醫學遺傳學重點實驗室,浙江溫州325035)
研究發現過量的重金屬銅對所有的生命體都有毒害作用[1]。隨著工農業的發展,許多含銅污染物被排放到環境中[2-4],對人們的生活與健康造成了巨大威脅,因此,監測與防治環境中重金屬銅的污染已不容忽視。檢測銅的傳統方法主要有原子吸收光譜法[5]和電感耦合等離子體質譜法[6]。但這些方法檢測成本高以及操作繁瑣,并且重金屬在環境中是以多種形態存在的,因此,通過檢測其總量難以客觀評價環境中重金屬的生物有效性與毒性效應[7]。而微生物報告菌株/全細胞微生物傳感器能夠有效彌補傳統方法的上述不足之處[8-9]。因為經過基因改造的工程菌株能以劑量依賴方式響應環境中特定化學物或一類化學物[10]。
本研究利用Red重組系統刪除了3 個銅離子(Cu2+)“外排泵”基因,獲得了對Cu2+高度敏感的E.coliΔcusA-ΔcopA-ΔcueO突變菌株,從而有利于微生物報告菌株富集水環境樣品中痕量Cu2+,有效提高了報告菌株的檢測靈敏度以及精確度。本研究構建的微生物報告菌株可與理化分析法互補分析水環境中銅的生物有效性及其總量,從而綜合評價水環境中銅的生物毒性效應。
1 材料和方法
1.1 材料 野生型E.coliMC4100 購于中國普通微生物菌種保藏管理中心;質粒pKD46、pKD3、pCP20、NGC-pMD19-T、pBAD-HisB和pET28a,菌種pET-28a/BL21、DH5α、NGC-T/DH5α和BL21均為浙江省醫學遺傳學重點實驗室保存;pMD19-T購自日本TaKaRa公司。
1.2 引物設計 根據Ecogene網站提供的目的基因及其啟動子與轉錄終止(CopAp、soxS、copA、cueO、cusA)序列及已知的gfpmut2基因序列,使用Primer5.0軟件設計引物(見表1-2),部分引物在基因的5′端或3′端引入酶切位點。
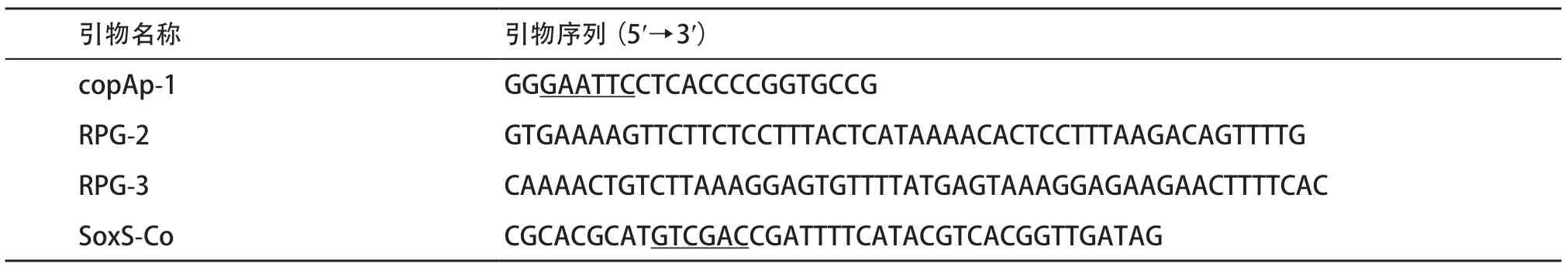
表1 用于構建報告基因的引物序列

表2 用于基因敲除和菌落PCR鑒定的引物序列
1.3 主要儀器 Milli-QAdvantageA10超純水系統(德國Millipore公司),HZQ-X100振蕩培養箱(哈爾濱東聯電子技術開發有限公司),DU-800核酸-蛋白分析儀(美國Beckman公司),電轉化儀(美國BIORAD公司),RF-5301熒光分光光度計(日本島津公司)。
1.4 方法
1.4.1 融合基因的制備與報告載體的構建:利用引物CopAp-1和RPG-2,以E.coli基因組作為模板擴增出具有融合位點的CopAp基因,利用引物RPG-3和soxSCo,以質粒NGC-pMD19-T作為模板擴增出具有相應融合位點的gfpmut2基因;將2個含有互相重疊位點的CopAp基因和gfpmut基因按照等摩爾的比例加入反應體系中作為模板,利用Pyrobest酶和ExTaq酶,以CopAp-1和SoxS-Co為引物進行PCR擴增(見圖1),PCR條件為:94℃4min;94℃30s,60℃30s,72℃3min,35 個循環;72℃10min;25℃5min。
擴增產物(CopAp::gfpmut2)經電泳檢測、切膠回收、加A反應后與pMD-19T載體連接,測序分析正確后再將其連接到表達載體pET28a上,獲得報告載體CopAp::gfpmut2-pET28a。將報告載體轉化DH5α后挑取陽性克隆提取質粒酶切鑒定,鑒定正確的克隆擴大培養后,用質粒提取試劑盒提取質粒,-20℃凍存備用,同時于-80℃菌種保存。
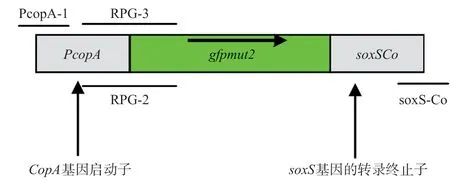
圖1 采用交叉PCR技術構建檢測Cu2+的報告基因
1.4.2 利用Red重組系統構建缺失copA、cueO 和cusA基因的Cu2+敏感菌株:以pKD3為模板,以各自敲除基因的引物PCR擴增含FRT位點(FLP重組酶識別位點)的氯霉素抗性基因(見圖2Step1),將其電轉化至含pKD46質粒的E.coliMC4100中(見圖2Step2)。pKD46/MC4100細菌感受態制備時經0.02%(m/v)阿拉伯糖誘導,使pKD46質粒在菌體內表達Gam、Bet和Exo3種λ噬菌體重組酶,其中Gam重組酶抑制大腸桿菌的RecBCD核酸外切酶V活性,使轉入氯霉素基因不被降解[11],同時在Exo和Bet重組酶輔助下氯霉素基因與3種待敲除基因copA、cueO、cusA的同源臂發生同源重組,從而置換出3個目的基因(見圖2Step3),獲得到copA::cat/MC4100、cueO::cat/MC4100和cusA::cat/MC41003種突變菌。然后將溫度敏感型質粒pCP20(42℃誘導FLP重組酶表達的同時質粒也逐漸丟失)[11]轉化至上述3種突變株感受態細胞中,消除FRT位點間的氯霉素抗性基因(見圖2Step4),分別得到copA、cueO和cusA基因缺失的單突變菌株。

圖2 采用Red重組系統進行基因敲除實驗
同樣操作,在單基因突變菌的基礎上制備雙基因突變菌及三基因突變菌。經PCR鑒定正確后共制備得到以下7種突變菌:單基因突變菌:ΔcopA、ΔcueO 和ΔcusA;雙基因突變菌:ΔcopA-ΔcueO、ΔcopA-ΔcusA 和ΔcueO-ΔcusA;三基因突變菌:ΔcopA-ΔcueO-ΔcusA。
1.4.3 含報告載體的微生物報告菌株的構建:將報告載體CopAp::gfpmut2-pET28a轉化至E.ColiMC4100和7種突變菌中,然后對每種細菌分別進行CopAp::gfpmut2目的基因PCR鑒定與突變菌PCR鑒定,確保重組質粒轉化成功。
1.4.4 野生菌和7 種突變菌及其對應的微生物報告菌株對Cu2+的敏感性測定:將野生菌株及7種突變菌株過夜培養,轉接至新鮮的M9基礎培養基中擴大培養2h左右,用M9基礎培養基將吸光度稀釋至OD600為0.005后分裝,加入Cu2+溶液,使其終濃度分別為0、0.125、0.25、0.5、1、2、4、8、16、32μmol/L,37℃、250r/min振蕩培養,檢測其10h內濁度改變,間隔2h測量各管菌液OD600值。重復實驗3次,每次操作3份平行管。
取8種含報告載體的宿主菌的過夜培養菌以新鮮LB培養基1:50稀釋并擴大培養至OD600為0.5左右,分裝,加入等體積不同濃度的銅標準溶液至各管菌液終濃度分別為1×10-10、1×10-9、1×10-8、1×10-7、1×10-6、1×10-5、1×10-4、1×10-3mol/L,另加等體積去離子水作為對照,每種濃度各做3份平行管。將菌液于37℃、250r/min振蕩培養2h后,取100μL菌液10 倍稀釋于0.9mLpH8.0Tris-HCl緩沖液中,加入34mg/mL氯霉素1μL(終濃度為34μg/mL),分別測量各管細菌稀釋液熒光強度及OD600值,并繪制曲線。
1.4.5 報告菌株檢測Cu2+的最佳實驗條件的優化:用移液槍吸取計算好的CopAp::gfpmut 2-pET28a/ΔcusA-ΔcopA-ΔcueO過夜菌種子液加入到50mL新鮮LB液體培養基中,使起始菌液OD600值約為0.02,于37℃、250r/min振蕩培養至OD600值分別為0.1、0.2、0.3、0.4、0.5和0.6,然后加入Cu2+溶液,使其終濃度為100μmol/L,另加等體積無菌MilliQH2O作為陰性對照。將菌液于37℃、250r/min振蕩培養4h后分別測量細胞熒光強度與濁度變化(為了使背景散射和內濾效應最小化,需要報告菌株的全細胞懸浮液的OD600≤0.3[12])。
此外,本研究也摸索了誘導時間對報告菌株檢測不同濃度Cu2+的靈敏度和線性范圍曲線擬合的影響。首先用移液槍吸取計算好的CopAp::gfpmut2-pET28a/ΔcusA-ΔcopA-ΔcueO過夜菌種子液加入到100mL新鮮LB液體培養基中,使起始菌液OD600值約為0.02,于37℃、250r/min震蕩培養至OD600值約為0.5,分裝(0.99mL/管),然后加入10μL不同濃度的Cu2+溶液至各管菌液,使其終濃度分別為10-7、5×10-7、10-6、5×10-6、10-5mol/L,另加等體積無菌MilliQH2O作為陰性對照。每種濃度各做9個平行管(每次取3個平行管)。將以上菌液于37℃、250r/min振蕩培養2、3、4h后,4000r/min水平離心10min,棄上清液,收獲的菌體重懸于1mLDB中,然后取50μL重懸菌液20倍稀釋于0.95mLDB中,混勻,分別測量其稀釋液熒光值與OD600值。實驗重復3次。
1.4.6 GFPmut2在微生物報告菌株中的誘導表達情況及鑒定:取CopAp::gfpmut2-pET28a/ΔcusAΔcopA-ΔcueO過夜培養菌以1:50擴大培養至OD600為0.5左右,分裝,加入等體積不同濃度的銅標準溶液至各管菌液,使終濃度分別為1.0×10-8、1.0×10-7、1.0×10-6、1.0×10-5、1.0×10-4、1.0×10-3mol/L,另加等體積去離子水作為空白對照。同時取pET28a/ΔcusA-ΔcopA-ΔcueO菌液作為陰性對照。將以上菌液于37℃、250r/min振蕩培養3h,收獲細菌,重懸于pH8.0Tris-HCl緩沖液中拍照。并選取銅濃度最高的誘導樣品,在激發波長為481nm條件下進行490~600nm范圍波長掃描,并用同樣的激發波長在激光共聚焦顯微鏡下觀察。
1.4.7 微生物報告菌株檢測Cu2+的特異性研究:選擇CopAp::gfpmut2-pET28a/ΔcopA菌株進一步研究其對其它金屬離子的響應情況,觀測其是否能被其他金屬離子啟動誘導表達GFPmut2蛋白。取CopAp::gfpmut2-pET28a/ΔcopA過夜培養菌以1:50擴大培養至OD600為0.5 左右,分裝后以終濃度為100μmol/L的不同二價金屬離子(Mn2+、Co2+、Ba2+、Fe2+、Ca2+、Mg2+、Zn2+、Cu2+、Pb2+、Cr2+)37℃誘導3h,分別取40μL菌液15倍稀釋于pH8.0Tris-HCl緩沖液中,測量熒光強度與OD600值。
1.4.8 流式細胞術分析報告菌株檢測信號的穩定性與可重復性:用移液槍吸取計算好的CopAp::gfpmut2-pET28a/ΔcopA-ΔcueO-ΔcusA過夜菌種子液加入到20mL新鮮LB液體培養基中,使起始菌液OD600值為0.02,加入10μLKana(100mg/mL),37℃、250r/min擴大培養至OD600為0.5左右,分裝(0.99mL/管),加入10μL不同濃度的Cu2+溶液至各管菌液,使其終濃度分別為1.0×10-8、1.0×10-7、1.0×10-6、1.0×10-5、1.0×10-4、1.0×10-3mol/L,另加等體積無菌MilliQH2O作為陰性對照。將以上菌液于37℃、250r/min振蕩培養3h后,4000r/min水平離心10min,棄上清液,收獲的菌體用經0.22μm無菌濾膜過濾的0.9%氯化鈉溶液(含有終濃度為20μg/mL氯霉素)重懸洗滌2~3次后,根據其OD600值將菌液濃度大致調為5×106CFU/mL,并于37℃震蕩孵育15min以獲得濃度均一的菌懸液。然后取1mL樣品移入流式管中。以激發光波長為488nm,發射光波長為505~545nm的檢測參數對樣本進行分析測試。儀器設定為每個樣本檢測10000個細胞。實驗重復3次。
1.4.9 確定報告菌株CopAp::gfpmut2-pET28a/ΔcopA-ΔcueO-ΔcusA 檢測Cu2+的線性范圍:取CopAp::gfpmut2-pET28a/ΔcopA-ΔcueO-ΔcusA過夜培養菌以1:50擴大培養至OD600為0.5左右,分裝,加入等體積不同濃度的銅標準溶液至各管菌中,使Cu2+終濃度分別為5.0×10-8、1.0×10-7、2.5×10-7、5.0×10-7、7.5×10-7、1.0×10-6、2.5×10-6、5.0×10-6、1.0×10-5mol/L,另加等體積去離子水作為對照,每種濃度各做3份平行管。將以上菌液于37℃、250r/min振蕩培養3h,取100μL菌液15倍稀釋于1.4mLpH8.0Tris-HCl緩沖液中,加入氯霉素使終濃度為34μg/mL,分別測量各管細菌稀釋液熒光強度及OD600值,并繪制曲線進行曲線擬合。
1.4.10 加標回收實驗:首先配制含MOPS緩沖劑的培養基:1g/L胰蛋白胨、0.5g/L酵母膏、1g/LNaCl,先加50mLMilliQH2O,振蕩至完全融解后加MilliQH2O定容至80mL,高壓蒸汽滅菌(121℃、30min),再加入10mL400mmol/L無菌MOPS緩沖液(pH7.2)[13],混勻。其次從溫州醫科大學茶山校區采集一個生活飲用水樣品,經0.22μm的無菌過濾器過濾。并在過濾后的生活飲用水中加入銅單元素標準品,使其終濃度分別為0.5和10mg/L,并采用原子吸收光譜法測定兩種水樣中銅含量。最后用移液槍吸取計算好的CopAp::gfpmut2-pET28a/ΔcopA-ΔcueO-ΔcusA過夜菌種子液加入到上述90mL含MOPS緩沖液的LB液體培養基中,使起始菌液OD600值約為0.02,37℃、250r/min震蕩培養至OD600值約為0.5,分裝(0.9mL/管),然后分別加入100μL上述含不同濃度Cu2+的生活飲用水,每種濃度各做3 個平行管。將以上菌液于37℃、250r/min振蕩培養3h后,4000r/min,水平離心10min,棄上清液,收獲的菌體重懸于1mLDB中,然后取50μL重懸菌液20倍稀釋于0.95mLDB中,混勻,分別測量其稀釋液熒光值與OD600值。然后根據標準曲線的回歸方程計算樣品中Cu2+的加標回收率。
2 結果
2.1 報告載體CopAp::gfpmut2-pET28a的構建 通過PCR擴增,得到大小為1500bp產物,與預期結果相符;陽性克隆質粒用EcoRI和SalI雙酶切鑒定,同樣也能切出1500bp的目的片段,目的片段序列經測序分析確證,表明報告載體CopAp::gfpmut2-pET28a構建成功(見圖3)。
2.2 copA、cueO和cusA基因缺失的Cu2+敏感菌株的構建 以CopA-No和CopA-Co、CueO-No和CueO-Co、CusA-No和CusA-Co為引物,分別對野生型E.ColiMC4100和通過基因敲除得到的7種突變菌進行PCR鑒定。如圖4所示,基因敲除后對應的PCR擴增片段約為1000bp,而未被敲除的菌株PCR擴增產物均與野生型菌PCR擴增產物條帶的大小一致。
2.3 野生菌及7種突變菌對Cu2+的敏感性 通過3次獨立重復實驗測定野生型E.coliMC4100以及7 種突變菌株對Cu2+的敏感性,得出8 種E.coli 各自的最小抑菌濃度(minimalinhibitoryconcentration,MIC)(見表3)。研究發現,培養10h后,由于培養基營養成分耗盡,部分細菌吸光度有所下降,故以0~8h作為細菌生長情況的監測時段。并且規定:8h后,使試驗管OD600值與對照管OD600值之比不大于10%的最低Cu2+濃度為細菌的MIC。
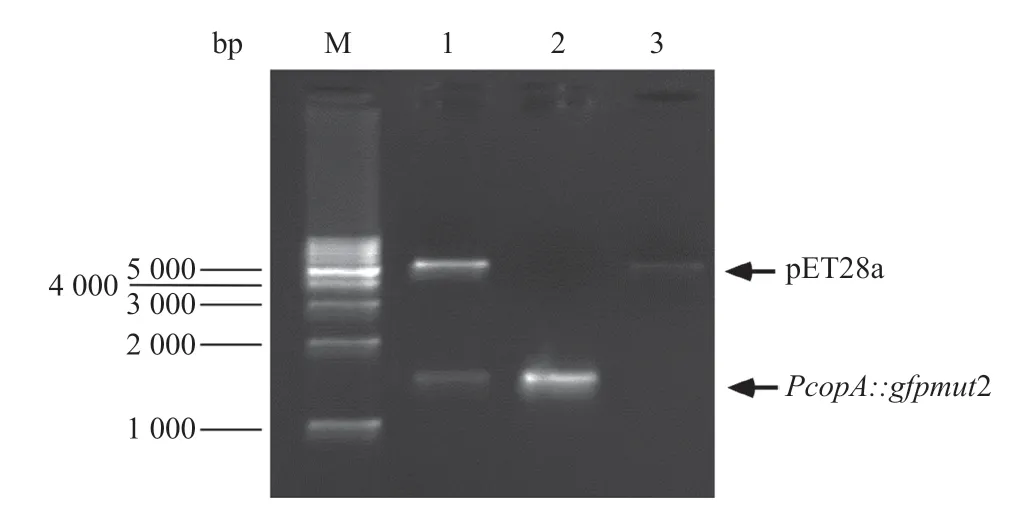
圖3 CopAp::gfpmut2-pET28質粒的雙酶切鑒定圖譜
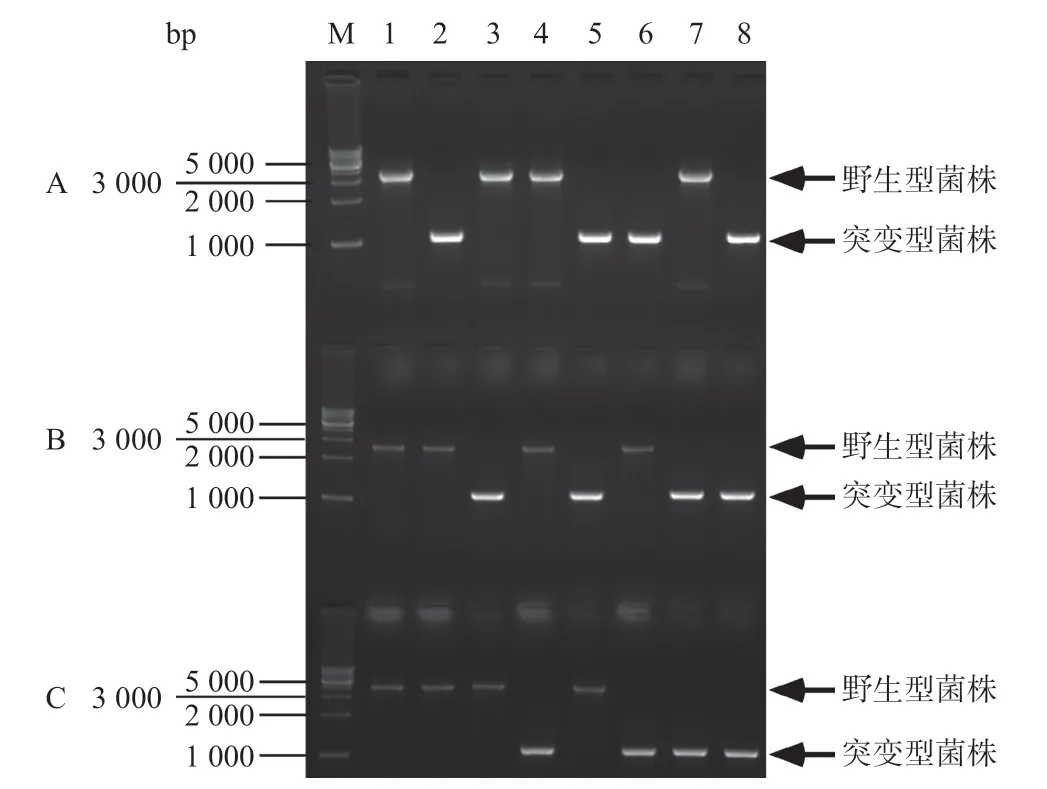
圖4 野生型E.coliMC4100及其7種突變菌株的菌落PCR鑒定圖譜
如表3所示,copA、cueO和cusA基因缺失的突變菌與野生型細菌相比,對Cu2+的敏感性均有一定程度的增加,其中以ΔcopA、ΔcopA-ΔcusA和ΔcopAΔcueO-ΔcusA對Cu2+最為敏感,其敏感程度均為野生型的64倍。由此推測,CopA在野生型E.coli 耐受Cu2+的機制中起主導作用。
2.4 鑒定Cu2+對微生物報告菌株細胞內熒光蛋白的誘導表達 以不同濃度的Cu2+誘導CopAp::gfpmut2-pET28a/ΔcusA-ΔcopA-ΔcueO菌株熒光蛋白的表達,肉眼可見細菌重懸液呈現明亮的綠色(見圖5A),且隨Cu2+濃度升高而顏色加深。其中圖5A中樣品8的發射光譜結果顯示在481nm激發光激發下在507nm處具有最大吸收峰(見圖5B),這些特征符合GFPmut2蛋白的熒光特征,這也證實了目的蛋白的表達;并且激光共聚焦顯微鏡圖譜顯示與10-4mol/LCu2+孵育后的整個報告菌株的菌體呈現亮綠色(見圖5C),更進一步驗證了Cu2+可以誘導熒光蛋白的表達。

表3 Cu2+對野生型E.coliMC4100及其7種突變菌株的MIC(μmol/L)

圖5 經Cu2+誘導后微生物報告菌株的表征圖譜
2.5 微生物報告菌株檢測水體中Cu2+最佳參數的優化 如圖6A-B所示,當初始細菌濁度在0.4~0.6時,加入100μmol/LCu2+與微生物報告菌株孵育4h后,微生物報告菌株的生長已進入平臺期,細胞熒光信號的累積以及熒光值與細菌濁度的比值同樣也進入平臺期,不再隨菌株濁度的變化而顯著變化。因此,本研究確定Cu2+誘導報告菌株累積熒光信號的最佳起始OD600值為0.5。
如圖6C所示,隨著與Cu2+孵育時間的延長,報告菌株熒光信號的累積與孵育時間呈正相關。但是孵育4h后,細胞中累積的相對熒光強度反而有所下降,這可能是由于細胞進入衰退期所致。經曲線擬合分析表明在3h時,在10-7至10-5mol/LCu2+濃度范圍內,報告菌株CopAp::gfpmut2-soxSCopET28a/ΔcusA-ΔcopA-ΔcueO累積的熒光信號強度與Cu2+濃度呈線性依賴關系,相關系數為0.99529。因此,本研究選擇3h為報告菌株與Cu2+最佳孵育時間。
此外,本研究也分析了水環境pH值對報告菌株細胞內增強型綠色熒光蛋白穩定性的影響。如圖6D所示,當水環境pH值小于6時,GFPmut2熒光信號顯著降低,在此pH值區間范圍內,熒光信號強度與pH值大小負相關;在pH值為6~8.5范圍內,GFPmut2發射的熒光信號強,且非常穩定。因此,本研究采用pH8.0Tris緩沖液重懸菌體后再進行熒光信號的檢測。并且規定加入LB培養基中水環境樣品的體積不能超過培養基體積的1/50,從而避免水環境樣品對培養體系pH值和滲透壓的顯著干擾。
2.6 微生物報告菌株CopAp::gfpmut2-pET28a/ΔcusA-ΔcopA-ΔcueO 對Cu2+的特異性分析 CopAp::gfpmut2-pET28a/ΔcopA報告菌株與終濃度為100μmol/L的不同二價金屬離子(Mn2+、Co2+、Ba2+、Fe2+、Ca2+、Mg2+、Zn2+、Cu2+、Pb2+、Cr2+)孵育3h后,測量各樣品的熒光及OD600值,分別將各金屬離子誘導的熒光/吸光度值與Cu2+誘導后的熒光/吸光度值(定義為100%)的百分比值為縱坐標繪制柱狀圖。如圖7所示,上述10種金屬離子,只有Cu2+能夠誘導報告菌株表達熒光蛋白,說明本研究構建的報告菌株具有很好的Cu2+響應特異性。
2.7 對比分析8種微生物報告菌株檢測Cu2+的靈敏性 根據8種微生物報告菌株對Cu2+的響應曲線的實驗結果表明,單獨缺失cueO或cusA,或者同時缺失cueO和cusA基因,對提高報告菌株檢測Cu2+的靈敏性,幾乎沒有顯著性影響。而缺失copA 基因的系列報告菌株對Cu2+的靈敏度顯著增強,尤其以ΔcusA-ΔcopA-ΔcueO三突變菌株為宿主菌的報告菌株對Cu2+的響應能力最強,產生的熒光信號也最強(見圖8)。綜上所述,本研究采用Red重組系統改造野生型E.coli 之后,可以顯著提高報告菌株的檢測性能。

圖6 微生物報告菌株檢測水體中Cu2+最佳參數的優化
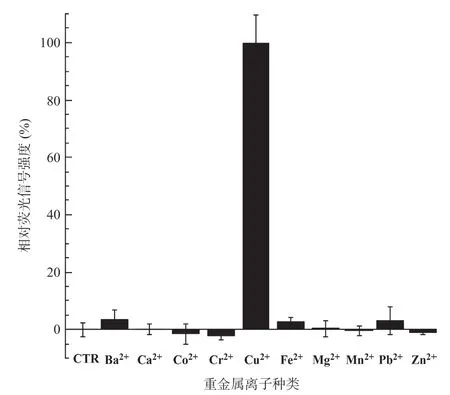
圖7 微生物報告菌株CopAp::gfpmut2-pET28/ΔcusA-ΔcopA-ΔcueO對Cu2+的特異性響應圖譜
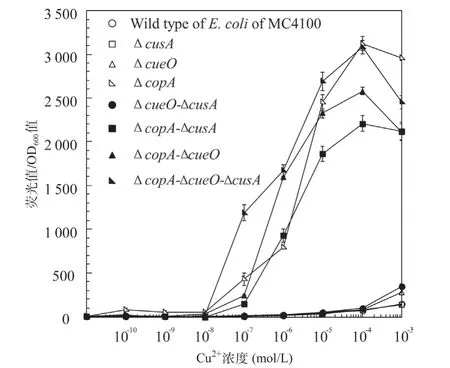
圖8 敲除參與Cu2+外排轉運基因對報告菌株響應Cu2+效果的影響
2.8 流式細胞術分析報告菌株熒光信號的穩定性與可重復性 TOM-PETERSEN等[14]研究表明,將Cu2+感應元件CopAp::gfpmut2插入到質粒載體中,會存在熒光本底值高和熒光信號不夠穩定的缺陷。但是,目前已有文獻報道的檢測Cu2+的微生物報告菌株,檢測元件都是位于質粒中[7,15]。因此,本研究采用流式細胞術分析了基于質粒的感應元件熒光信號的穩定性與可重復性。如圖9所示,雖然與不同濃度Cu2+孵育后報告菌株的單細胞熒光峰圖在左邊低熒光值區域具有明顯拖尾現象,甚至無熒光細胞數明顯增加(見圖9B-C)。但是相對沒有添加外源性Cu2+的陰性對照(見圖9A),隨著Cu2+濃度的增加,熒光值峰圖明顯右移,高熒光值區域的熒光峰圖離散度明顯變小(見圖9B-C)。尤其是圖9D表明即使是獨立重復實驗,報告菌株應答微摩爾濃度以上Cu2+產生的熒光信號,具有很好的可重復性(SD值<20%)。

圖9 流式細胞術分析報告菌株熒光信號的穩定性與可重復性
2.9 CopAp::gfpmut2-pET28/ΔcopA-ΔcueO-ΔcusA報告菌株檢測Cu2+的線性范圍 以等體積終濃度分別為5.0×10-8、1.0×10-7、2.5×10-7、5.0×10-7、7.5×10-7、1.0×10-6、2.5×10-6、5.0×10-6、1.0×10-5mol/L的Cu2+誘導CopAp::gfpmut2-pET28/ΔcopA-ΔcueO-ΔcusA報告菌株表達綠色熒光蛋白,測量其熒光強度和吸光度,以Cu2+終濃度的對數值為橫坐標,熒光強度/吸光度值為縱坐標進行曲線擬合(見圖10),其回歸方程為y=14460+1924.8log(x),相關系數R2=0.99477,表明在0.05~2.5μmol/L(0.0032~0.16mg/L)Cu2+濃度范圍內,CopAp::gfpmut2-pET28/ΔcopA-ΔcueO-ΔcusA報告菌株產生的熒光信號與Cu2+濃度之間具有很好的線性關系。參考我國各類國標中針對銅的限制表明,CopAp::gfpmut2-pET28/ΔcopA-ΔcueO-ΔcusA報告菌株具備監測地表水和地下水環境中痕量Cu2+的應用潛力(見表4)。
2.10 加標回收實驗 為了進一步評估報告菌株應用于環境水樣的潛力,本研究在生活飲用水(采集于溫州醫科大學茶山校區)中加入已知濃度Cu2+,分別采用微生物報告菌株和原子吸收光譜法進行檢測,實驗結果表明微生物報告菌株對生活飲用水中0.0496mg/L和0.9634mg/LCu2+的回收率分別為87.90%和104.37%(見表5),而我國針對生活飲用水衛生標準(GB5749-2006)中銅的限值為1mg/L,針對污水綜合排放標準(GB8978-1996)的一級標準限值為0.4mg/L(見表4)。因此,本研究所述微生物報告菌株的靈敏度完全具備應用于監測生活飲用水和污水中Cu2+污染的潛力。
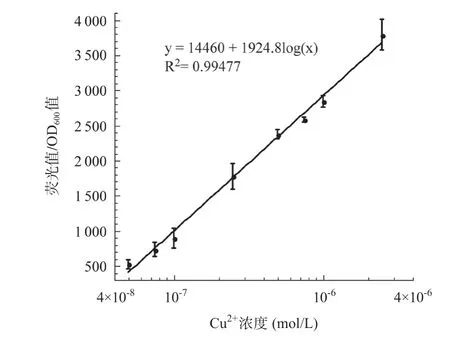
圖10 CopAp::gfpmut2-pET28/ΔcopA-ΔcueO-ΔcusA報告菌株檢測Cu2+的線性范圍

表4 我國各類國標中針對銅的限制

表5 報告菌株CopAp::gfpmut2-pET28/ΔcopA-ΔcueO-ΔcusA檢測生活飲用水樣品中Cu2+的加標回收率
3 討論
在負責維持E.coli 體內Cu2+穩態的cop操縱子中,由于cusA、copA和cueO3個結構基因對Cu2+的外排作用,導致E.coli 胞質內Cu2+濃度不會隨外環境Cu2+的增加而升高,從而限制了以其為宿主菌的報告菌株的檢測靈敏性,并且也會導致野生型報告菌株對Cu2+耐受,進一步降低其檢測靈敏度。為了解決上述問題,本研究首先對野生型E.coli 宿主菌進行了基因工程改造:①刪除了E.coli 中負責將Cu2+從細胞內向細胞外泵出的copA和cusA基因以及負責將Cu+氧化為Cu2+,防止外排到周質中Cu+再次進入胞質的cueO基因;②以熒光強度更強的增強型綠色熒光蛋白GFPmut2[16-17]作為報告標志基因構建融合報告載體。研究結果表明本研究構建的ΔcopAΔcueO-ΔcusA突變菌對Cu2+非常敏感,在M9基礎培養基中其敏感性是野生型E.coliMC4100 的64 倍(見表3)。雖然相對ΔcopA 單基因缺失菌株,三突變菌株的敏感性并未顯著增加,但是相對E.coliMC4100,ΔcueO-ΔcusA雙突變菌對Cu2+的敏感性提高了4倍。說明在刪除copA基因的基礎上面,進一步刪除cueO 和cusA基因是有助于提高報告菌株對Cu2+敏感性的。并且從8種微生物報告菌株對Cu2+的響應曲線可以看出(見圖7),相對野生型報告菌株,宿主菌為ΔcueO、ΔcusA和ΔcueO-ΔcusA的報告菌株對Cu2+的敏感性略有提高;而含有copA基因缺失的報告菌株對Cu2+的敏感性顯著增強,該研究結果與文獻報道一致[15];尤其是E.coliΔcopA-ΔcueOΔcusA三基因突變菌響應Cu2+的熒光信號最強,線性范圍最好,其累積的熒光信號強度為野生型的40倍左右(見圖8)。綜上所述,本研究構建的基于E.coliΔcopA-ΔcueO-ΔcusA 三基因突變菌的報告菌株,相對基于野生型E.coliMC4100或者ΔcopA單突變菌株,能夠有效提升CopAp::gfpmut2檢測元件對水溶液中鈉摩爾數量級Cu2+的響應能力。
綠色熒光蛋白(greenfluorescentprotein,GFP)因具有檢測方便、熒光穩定、無毒害、通用性、不需要外源性底物、輔助因子和ATP、易于實現融合表達以及在活細胞內定時定位觀察等優點,已成為微生物報告菌株中最常用報告基因之一[18]。本研究所選報告基因為增強型GFPmut2,它是在野生型GFP中突變3個氨基酸位點(S65A、V68L、S72A)[17],它不僅具有更強的熒光信號,而且只需日光中的藍光(481nm)就可以激發出亮綠色的熒光,這樣不僅可以提高報告菌株的監測靈敏度,更重要的是不需要紫外設備就能通過肉眼觀測菌體顏色變化判斷目標檢測物濃度高低,這為野外實時監測帶來很大方便。本研究實驗數據表明,經Cu2+誘導后,通過肉眼即可觀察到報告菌株菌體呈亮綠色,且具有濃度依賴性(見圖5A)。
目前,已有多種基于cop操縱子的Cu2+檢測元件被文獻報道,比如copAp::luxCDABE(檢測Cu2+的線性范圍為3~30μmol/L)[19],pSLcueR/copAp::luxCDABE(對Cu2+的最低檢出限值(limitofdetection,LOD)為0.15μmol/L)[23],pSL-cueR/copAp::PpyWT(LOD:0.15μmol/LCu2+)[21]和pPROBEcueR/cueAp::gfp感應元件(檢測Cu2+范圍為1~70mg/L)[7],這些感應元件都被插入到質粒載體中,然后再轉化到野生型E.coli 或者惡臭假單胞菌中,從而構建特異性檢測水體中生物有效性Cu2+的全細胞微生物報告菌株。此外,KANG等[15]最新研究表明,在E.coliΔcopAΔcueR 雙突變菌中共表達pCopAp-eGFP和pCDF-Duet-CueR質粒,可以顯著提高報告菌株檢測Cu2+的敏感性,其檢測Cu2+范圍為0~10μmol/L。本研究基于E.coliΔcopA-ΔcueOΔcusA三基因突變菌構建的報告菌株檢測Cu2+的線性范圍為0.05~2.5μmol/L(0.0032~0.16mg/L),相對上述文獻報道的同類微生物報告菌株,其檢測靈敏度具有明顯的優勢。
此外,由于環境樣品成分復雜,可能會影響報告菌株的生長和GFPmut2蛋白的表達。比如,固體顆粒或樣品顏色可能會增強/猝滅報告菌株GFPmut2蛋白熒光,有毒物質(比如有機磷殺蟲劑或各類除草劑)的抑制效應或營養物質的促進效應[22]。因此,本課題組在后續實驗中需要構建組成性表達GFPmut2的內參菌株[20,23],以校正上述因素對報告菌株熒光表達的影響。在每次實驗中,將同時使用報告菌株和內參菌株對環境樣品進行檢測。內參菌株被用來確定校正系數[20]。
綜上所述,本研究構建的突變菌有很好的線性范圍,跟同類報告菌株相比具有更高的靈敏度。同時采用增強型GFP作為報告基因,能通過肉眼就能觀測菌體顏色的變化,從而判斷目標檢測物濃度的高低,具有實時監測的優勢。當然我們今后將繼續對報告菌株CopAp::gfpmut2-pET28/ΔcopA-ΔcueOΔcusA進行改進與優化,并將其構建為能夠檢測水體中Cu2+的微生物傳感器,制定標準化的檢測流程,并應用于水體環境中Cu2+的檢測與監測。為進一步研發能夠實時、同步、快速檢測多種重金屬污染物(包括鉻、鉛、汞、銅等)的微生物芯片傳感器提供技術上的借鑒和良好的應用范例。
猜你喜歡
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
南方人物周刊(2017年32期)2017-10-28 22:48:36
南風窗(2016年26期)2016-12-24 21:48:09
海峽科技與產業(2016年3期)2016-05-17 04:32:12
南風窗(2015年22期)2015-09-10 07:22:44
南風窗(2015年14期)2015-09-10 07:22:44
