特發(fā)性偏身肥大癥合并腦白質(zhì)病變1例報(bào)告
2020-05-12 02:42:30王曉玉郭雯琪王迎新
中風(fēng)與神經(jīng)疾病雜志 2020年4期
王曉玉, 郭雯琪, 曹 華, 王迎新
特發(fā)性偏身肥大癥(idiopathic hemihypertrophy,IH)是一種先天罕見(jiàn)疾病,以一側(cè)肢體、軀干和頭面部或者整個(gè)半身不對(duì)稱生長(zhǎng)、肥大為主要特征,肥大包括軟組織以及骨骼[1,2]。偏身肥大癥(hemihypertrophy,HH)可以獨(dú)立存在,也可以是某些先天性異常綜合征的一部分,前者稱為IH[1,2]。該病發(fā)病率極低,為1/13000~1/86000[3,4],臨床醫(yī)生對(duì)此疾病認(rèn)識(shí)少,患者就醫(yī)困難。現(xiàn)報(bào)道1例IH合并腦白質(zhì)病變患者。
1 臨床資料
患者,男,27歲。因“發(fā)現(xiàn)右下肢增粗17 y” 于2019年5月21日入住大連醫(yī)科大學(xué)附屬第一醫(yī)院神經(jīng)內(nèi)科。患者17 y前10歲時(shí)發(fā)現(xiàn)雙下肢長(zhǎng)度不等,右下肢較左下肢長(zhǎng)約1 cm,此后發(fā)現(xiàn)右下肢直徑較左下肢增粗,15歲到18歲期間右下肢增粗明顯。多次于外院就醫(yī),未明確診斷。無(wú)肢體疼痛、麻木、無(wú)力、二便障礙。無(wú)抽搐、無(wú)意識(shí)喪失。既往史:體健。個(gè)人史:為父母第2胎,孕期正常,母親無(wú)放射線及毒物接觸史。足月順產(chǎn),出生時(shí)體重3.2 kg,1歲半會(huì)走路。幼時(shí)經(jīng)常跌倒,運(yùn)動(dòng)能力較同齡人略差。智力正常,理工科大學(xué)本科畢業(yè),工程師。未婚。家族史:父母非近親結(jié)婚,有一姐姐體健。家族中無(wú)同類(lèi)患者。查體:頸、胸、背部多處可見(jiàn)皮下結(jié)節(jié),質(zhì)軟,不可移動(dòng)。胸部劍突水平皮膚見(jiàn)直徑5 cm類(lèi)圓形皮膚色素沉著。雙手掌大小魚(yú)際肌較平,掌紋深,雙肘屈曲側(cè)皮紋少、淺。右下肢較左下肢長(zhǎng)2 cm(R:105 cm;L:103 cm);右下肢周徑較左下肢明顯增粗:雙下肢膝以上10 cm周徑:右側(cè)較左側(cè)圓周長(zhǎng)11.5 cm(R:57.5 cm;L:46 cm);雙下肢膝部周徑:右側(cè)較左側(cè)圓周長(zhǎng)16.5 cm(R:54.5 cm;L:38 cm);雙下肢膝以下10 cm周徑:右側(cè)較左側(cè)圓周長(zhǎng)8 cm(R:47 cm;L:39 cm);腳踝周徑:右側(cè)較左側(cè)長(zhǎng)5.5 cm(R:30 cm;L:24.5 cm)。右膝關(guān)節(jié)表面皮紋淺,雙足輕度馬蹄內(nèi)翻足,右足皮膚粗糙、脫屑、體毛重(見(jiàn)圖1)。神經(jīng)系統(tǒng)查體:神清,語(yǔ)明。精神、智能正常。顱神經(jīng)查體未見(jiàn)明顯異常。右側(cè)L2~S2水平痛覺(jué)減退。雙上肢及右下肢腱反射減弱。右側(cè)Babinski征(+),左側(cè)Babinski征(-),雙側(cè)Chaddock征(+)。其余神經(jīng)系統(tǒng)查體未見(jiàn)明顯異常。輔助檢查:頭部MRI+MRA:雙側(cè)額頂葉皮質(zhì)及皮質(zhì)下異常信號(hào),T2Flair斑片狀高信號(hào)(見(jiàn)圖2);頭部MRA未見(jiàn)異常。頸椎MRI:頸5/6椎間盤(pán)輕度膨出;胸椎MRI未見(jiàn)異常。腰椎MRI:腰5骶1椎間盤(pán)突出。 雙下肢股骨、脛腓骨MRI:右側(cè)大腿肌肉脂肪化,右側(cè)小腿前內(nèi)側(cè)皮下脂肪增厚。右側(cè)股骨、脛骨直徑較左側(cè)增粗(見(jiàn)圖1)。肌電圖:右下肢未見(jiàn)明顯神經(jīng)源性或肌源性損害。腦電圖:界限性腦電地形圖,界限性腦電圖,背景快波活動(dòng)增多。心電圖:正常。胸部CT(平掃+HR):雙肺多發(fā)磨玻璃結(jié)節(jié),雙肺下葉少許炎癥改變。腹部彩超、泌尿系彩超未見(jiàn)異常。心臟彩超、頸部大血管彩超、雙下肢動(dòng)脈、深靜脈彩超未見(jiàn)明顯異常。化驗(yàn)檢查:血、尿、便常規(guī);肝、腎功能;血糖、血脂、電解質(zhì)、肌酸激酶、心肌標(biāo)志物、血沉;肝炎病毒系列、梅毒、艾滋病毒;風(fēng)濕、免疫、腫瘤標(biāo)志物均未見(jiàn)明顯異常。頸部皮膚結(jié)節(jié)切除行皮膚病理染色:表皮囊腫伴肉芽腫性炎癥。遺傳病全外顯子組基因測(cè)序(金域醫(yī)學(xué)):檢測(cè)到一個(gè)雜合基因變異:9q34 TSC1 Intron10 c.1029+3 A>G,為內(nèi)含子區(qū)微小點(diǎn)突變,臨床意義未明。其父親、母親均未檢測(cè)到該雜合基因突變。
2 討 論
IH是一種先天罕見(jiàn)疾病,以一側(cè)肢體、軀干和頭面部或者整個(gè)半身不對(duì)稱生長(zhǎng)、肥大為主要特征,肥大包括軟組織以及骨骼[1,2]。其潛在的病理機(jī)制是肥大組織細(xì)胞增殖速率增高,而不是細(xì)胞體積增大[3,4]。HH可以獨(dú)立存在,稱為IH,也可以是某些先天性異常綜合征的一部分[1~7]。該病發(fā)病率極低,大約為1/13000~1/86000[3,4]。多毛癥(hypertrichosis)是指在與同種族、同性別、同年齡個(gè)體相比毛發(fā)過(guò)度生長(zhǎng),多毛癥可以分布于全身,也可以局限于一小片,目前認(rèn)為先天性多毛癥是一種遺傳性疾病或是散發(fā)性基因突變的結(jié)果。迄今為止,全世界僅有4例IH合并偏身多毛癥(hemihypertrichosis)的病例報(bào)告[2]。因此,臨床醫(yī)生對(duì)此疾病認(rèn)識(shí)少,本例患者在就診于我科之前多次輾轉(zhuǎn)于各醫(yī)院門(mén)診都沒(méi)有得到確診,直到27歲才得以明確診斷,這其實(shí)面臨著極大的疾病風(fēng)險(xiǎn),因?yàn)镠H患者在兒童與少年時(shí)代易患兒童性惡性腫瘤(如Wilms腫瘤、肝母細(xì)胞癌、神經(jīng)母細(xì)胞瘤和腎上腺癌等[1~7]),需要定期進(jìn)行腫瘤監(jiān)測(cè)。此例患者入院后經(jīng)過(guò)系統(tǒng)檢查未發(fā)現(xiàn)惡性腫瘤,僅合并肥大側(cè)肢體體毛重,因此診斷為特發(fā)性偏身肥大癥合并偏身多毛癥(idiopathic hemihypertrophy associated with hemihypertrichosis)。另外,此前報(bào)告的4例患者3位為嬰幼兒時(shí)期就診,1例16歲時(shí)就診,4位患者就診時(shí)頭部MRI檢查無(wú)陽(yáng)性發(fā)現(xiàn),本例27歲男患雖無(wú)中樞神經(jīng)系統(tǒng)癥狀,但在查體時(shí)發(fā)現(xiàn)有雙側(cè)錐體束受損體征,頭部MRI T2Flair顯示雙側(cè)額頂葉皮質(zhì)及皮質(zhì)下斑片狀高信號(hào)病灶。該患者查體發(fā)現(xiàn)的神經(jīng)系統(tǒng)受累體征可以以額頂葉皮質(zhì)及白質(zhì)病變解釋。該患者頸、胸、背部多處可見(jiàn)的皮下結(jié)節(jié)經(jīng)取頸部皮下結(jié)節(jié)活檢為表皮囊腫伴肉芽腫性炎癥。
包含偏身肥大癥狀的常見(jiàn)先天綜合征包括:Beckwith-Wiedemann綜合征(Beckwith-Wiedemann Syndrome,BWS)、Klippel-Trenaunay-Weber綜合征以及Proteus綜合征等[1~7]。BWS以偏身肥大、內(nèi)臟肥大、巨大畸形新生兒、巨舌、臍疝和胚胎來(lái)源的腫瘤為主要表現(xiàn)[1,2,4],80%以上的患者發(fā)現(xiàn)染色體11p15.5分子異常,即父系來(lái)源的單親二體性(paternal uniparental disomy,pUPD 11p15)[4]。Klippel-Trenaunay-Weber綜合征以肢體或手足軟組織肥大、葡萄酒色痣和靜脈曲張畸形為主要臨床表現(xiàn),與VG5Q基因激活物突變有關(guān)[1,2]。Proteus綜合征表現(xiàn)為長(zhǎng)骨肥大,局部尤其是手足巨大畸形、足底畸形生長(zhǎng),血管瘤、脂肪瘤、線狀疣狀表皮痣和局部皮膚發(fā)育不全,相關(guān)致病基因未明[1,4]。與HH相關(guān)的基因缺陷由Martin等[8]于2005年首先報(bào)告。Bliek等對(duì)73例HH患者進(jìn)行了表觀遺傳學(xué)分析,15%的HH患者檢出pUPD 11p15缺陷;6%患者檢出KGNQ1OT1去甲基化;約3%患者檢出H19過(guò)甲基化[5]。
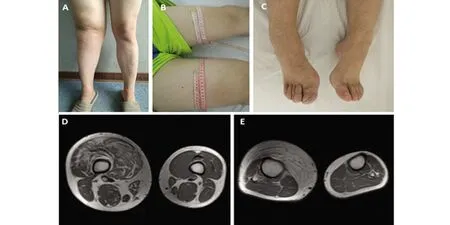
A:右下肢較左下肢明顯增粗;B:膝以上10 cm周徑:右側(cè)57.5 cm,左側(cè)46 cm;C:右足較左足大,右足背體毛重于左足;D:雙側(cè)股部MRI示右股部周徑大于左股部,右股骨直徑大于左股骨,右股部肌肉脂肪化;E:雙小腿MRI示右小腿周徑大于左小腿,右脛骨直徑大于左脛骨,右小腿前部皮下脂肪明顯增生
圖1 患者雙下肢外觀、雙股部與雙小腿MRI橫斷面

圖2 患者頭部MRI T2 Flair序列:A、B、C顯示雙側(cè)額葉、頂葉皮質(zhì)及皮質(zhì)下白質(zhì)散在高信號(hào)改變
HH被認(rèn)為是由馬賽克式(a mosaic form)的基因缺陷導(dǎo)致的馬賽克式的過(guò)度生長(zhǎng),實(shí)際上,研究發(fā)現(xiàn)pUPD總是以馬賽克形式存在,增大的組織器官中由較大比例的二體性細(xì)胞(disomic cells)組成[5]。Itoh 等[9]研究1例BWS不同組織甲基化形式,他們展示了肥大器官含有pUPD的細(xì)胞比例較高(40%~50%),pUPD也以較低百分比存在于血液淋巴細(xì)胞中,可以通過(guò)常規(guī)檢測(cè)做出診斷。當(dāng)pUPD僅以很低的比例存在于細(xì)胞中,很可能不存在于血液淋巴細(xì)胞中。Shuman等[10]設(shè)想對(duì)于一部分不能解釋的IH病例pUPD僅存在于肥大組織中,因此,在常規(guī)的血液診斷性檢測(cè)中不能被檢測(cè)到。
該患者外周血基因檢測(cè)未查到pUPD 11p15。所查到的9q34 TSC1 Intron10 c.1029+3 A>G,為內(nèi)含子區(qū)微小點(diǎn)突變,臨床意義未明。其父親、母親均未檢測(cè)到該雜合基因突變。患者右下肢肥大不影響生活與工作,目前無(wú)需特殊治療,其神經(jīng)系統(tǒng)體征及頭部MRI病變我們將長(zhǎng)期隨訪。
