線粒體神經胃腸型腦肌病1例報告并文獻復習
2020-05-12 02:42:28張小偉盧麗君李新明鄧幼清吳華東
中風與神經疾病雜志 2020年4期
張小偉, 盧麗君, 楊 珍, 李新明, 鄧幼清, 吳華東
線粒體腦肌病(mitochondrial encephalomyopathy,ME)是由于線粒體DNA(mitochondrial DNA,mtDNA)或核DNA(nucleus DNA,nDNA)缺陷導致的線粒體結構和功能障礙,使ATP生成不足而引起的中樞神經系統和肌肉疾病。線粒體遺傳病是近40多年來發現的一個新疾病體系,該組疾病臨床表現復雜多樣,確診率不高,容易誤診,現報道1例臨床罕見的特殊類型的線粒體腦肌病-線粒體神經胃腸型腦肌病,并復習文獻,以提高診斷準確率,減少誤診。
1 病例資料
患者,男性,33歲,農民,因上瞼下垂、全身乏力5 y余,加重1 y半入我院神經內科。患者緣于5 y前無明顯誘因開始出現上瞼下垂、全身乏力,易疲勞,能獨立行走,能上樓梯,伴有記憶力減退,近記憶力差為主,無意識障礙、頭痛、頭暈;無惡心、嘔吐;無胸悶、胸痛;無肢體麻木、無明顯肌肉萎縮等不適,未予重視。近1 y半感上述癥狀加重,尚能平穩行走,能上樓梯,但行走較前遲緩,且反應遲鈍,有腹瀉,約6次/d,多則每天10余次,有時解黃色水樣便,體重下降約10 Kg,無肢體抽搐、肌肉疼痛,無小便障礙等,曾于半年前在當地縣人民醫院診治,診斷為“胃腸功能紊亂”,后感癥狀持續存在,且漸加重,活動后明顯。既往3歲時曾有白喉病史,治療后好轉。5歲~8歲發熱時有抽搐病史,抽搐時有眼球上翻、牙關緊閉,持續5~6 min后可自行緩解,1 y發作1~2次。否認藥物及毒物嗜好史。否認有遺傳病家族史,家族成員無類似癥狀。入院查體:體型呈無力型(見圖1),營養不良,心肺腹未見明顯異常。神經系統查體:神清,構音欠清,右利手,記憶力、計算力、理解力減退,雙眼瞼下垂,兩側瞳孔等大等圓,直徑約3.5 mm,對光反射靈敏,雙眼球上下左右運動均受限(水平運動受限明顯),無眼球震顫,兩側額紋對稱,雙側鼻唇溝等深,口角無歪斜;雙耳聽力粗測正常,兩側軟腭上抬可,咽反射存在,洼田飲水試驗正常,伸舌居中,四肢肌力5-級,雙側肢體肌張力正常;雙側指鼻試驗及跟膝脛試驗笨拙,閉目難立征正常;感覺檢查正常;四肢腱反射對稱減弱,病理征未引出;腦膜刺激征陰性。疲勞試驗陽性。輔助檢查:血常規、尿液分析、大便分析+潛血、凝血功能、肌酸激酶、肌酸激酶同工酶MB、電解質、肝腎功能、血糖、糖化血紅蛋白、甲五聯、梅毒螺旋體抗體、人類免疫缺陷病毒抗體均正常。腫瘤標志物檢測(男):細胞角蛋白19片段測定:4.56 ng/ml(0.1~3.3 ng/ml),余指標正常。血脂:載脂蛋白A1:198.5 mg/dl(107~177 mg/dl)、載脂蛋白B:43.7 mg/dl(60~138 mg/dl)、甘油三酯:0.52 mmol/L(0.56~1.70 mmol/L),余指標正常。大便培養+真菌培養:未見明顯異常。靜息狀態血乳酸3.2 mmol/L、平路步行5 min后血乳酸4.3 mmol/L、上下樓5 min后血乳酸5.0 mmol/L(0.7~2.1 mmol/L)。頭部MRI平掃未見明顯異常(見圖2)。頭部磁共振波譜(MRS):未見明顯異常乳酸(Lac)峰(見圖3)。肌電圖:廣泛神經源性損害。重復電刺激:斜方肌高頻刺激見遞減現象,眼輪匝肌、小指展肌刺激未見遞減現象。腦干聽覺誘發電位:雙側聽覺通路傳導正常。眼底照相:雙眼底未見明顯異常。動態腦電圖:異常腦電圖(雙側前額部癇樣放電)。日常生活能力量表測定:功能有一定程度的下降。簡易智力狀態檢查量表測定:有癡呆表現。長谷川癡呆量表測定:可能有癡呆。心電圖:竇性心律,偶發房早。肌肉(左肱二頭肌)活檢:HE染色見肌纖維大小輕度不等,可見較多小圓纖維和角形纖維,輕度核內移、肌分裂,并可見散在空泡肌纖維,空泡較大,分布于肌纖維膜下,未見鑲邊空泡(RV),未見明顯壞死及再生纖維,未見明顯炎性細胞浸潤,肌內衣、肌束衣輕度增生(見圖4~圖6);Gomori染色見大量不整紅邊纖維(RRF)(見圖7、8);ORO染色見較多脂滴明顯增多肌纖維(見圖9);PAS染色見糖原明顯增多肌纖維(見圖10);NADH染色見明顯深染肌纖維(見圖11);肌纖維內及血管周圍未見明顯CD8陽性、CD68陽性細胞浸潤,符合線粒體病的肌肉病理改變。結合病史及病理結果,診斷線粒體神經胃腸型腦肌病,給予復合維生素B、輔酶Q1O及左卡尼汀口服,經治療10 d患者解大便次數減少, 其它癥狀無明顯改善,帶口服藥出院。
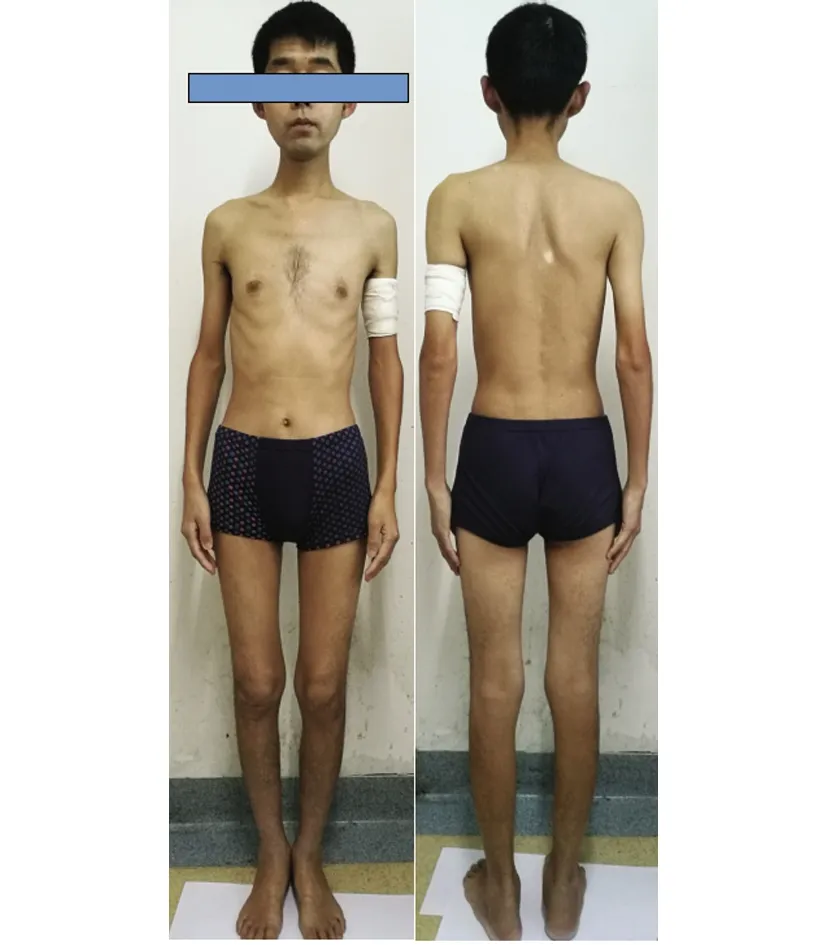
圖1 患者體型瘦長
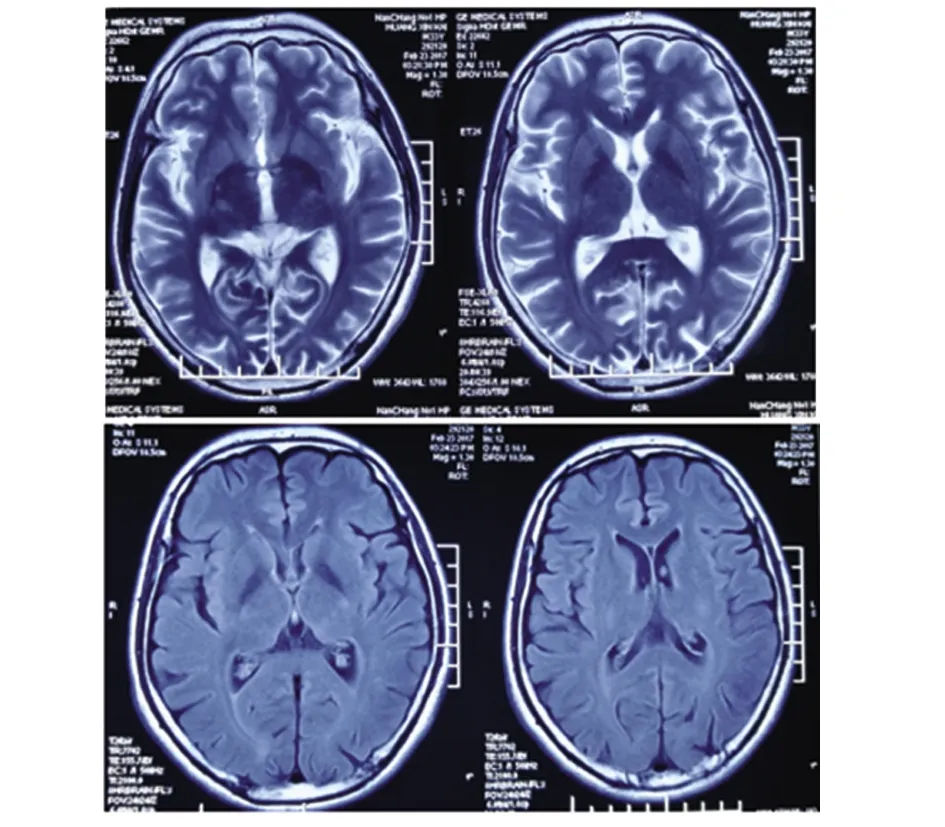
圖2 患者頭部MRI T2序列和FLAIR序列均未見異常信號改變
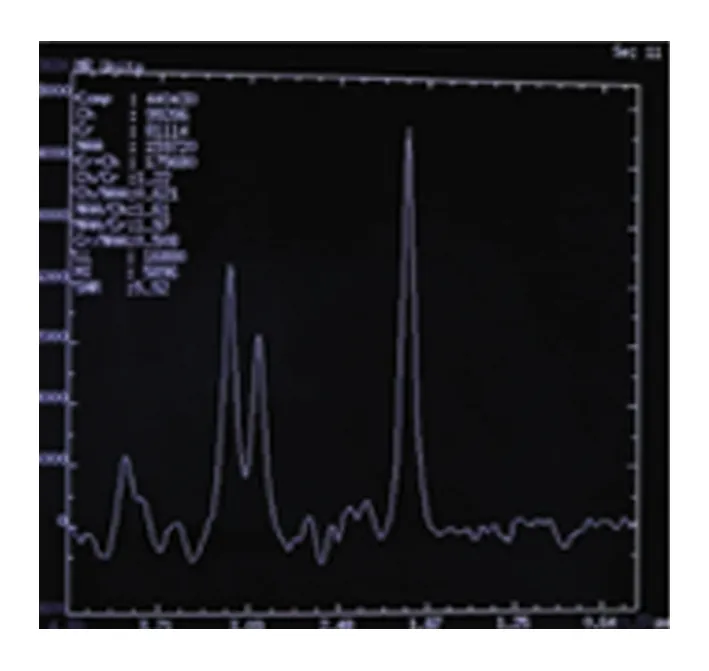
圖3 患者頭部MRS成像未見異常乳酸(Lac)峰
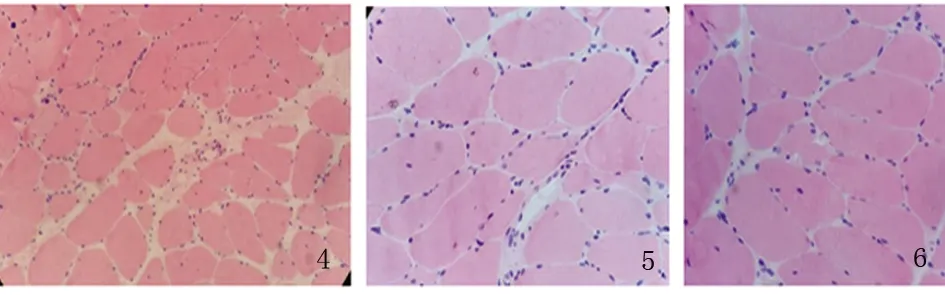
圖4 HE染色×200;圖5 HE染色×400;圖6 HE染色×400
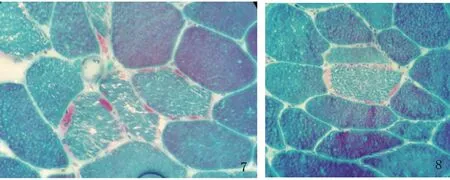
圖7 Gomori染色×400;圖8 Gomori染色×400
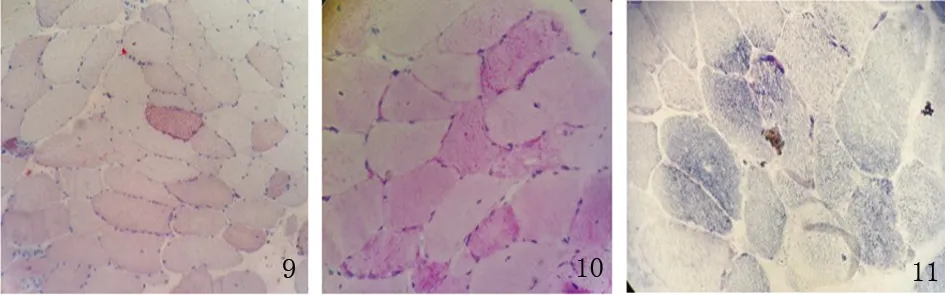
圖9 ORO染色×200;圖10 PAS染色×400;圖11 NADH染色×400
2 討 論
線粒體腦肌病是一種母系遺傳的代謝性疾病,因線粒體結構和功能異常所致的以腦和肌肉受累為主的多系統疾病。Cohen等[1]根據該病的臨床表現、病理特點將線粒體腦肌病分為7個亞型,也有文獻報道各亞型的癥狀可彼此重疊[2]。
線粒體神經胃腸型腦肌病(MNGIE)在1976年由Okamura等[3]首次報道,該疾病發病罕見,國內報道次數極少,該疾病從嬰兒到中年均可發病,多數患者在青少年或成年早期起病。許二赫等[4,5]在國內首次報道MNGIE并系統回顧該疾病多表現為胃腸運動障礙及惡病質、眼肌麻痹、周圍神經損害、腦白質病變以及其它癥狀,如聽力異常、自主神經功能紊亂、貧血、身材矮小等5種類型。此外,屈雪萍等[6]表明MELAS在實驗室檢查中多出現血乳酸運動試驗和丙酮酸水平升高;頭部MRI可表現為“花邊征”或“飄帶征”。肌肉活檢可見破碎紅纖維(RRF)、SDH染色增強的纖維或線粒體超微結構異常。
mtDNA基因檢測可表現為A3243G、T3271C、A3252G位點基因突變,其他MELAS的點突變還包括tRNAVal、tRNAPhe、tRNALy、tRNAHis等。MNGIE目前尚無統一的診斷標準,多從患者的特征性臨床表現及神經電生理表現下臨床診斷,基因檢測有助于該疾病診斷。本病例患者雖未做基因檢測,但符合該疾病的特征性臨床表現及神經電生理表現。目前本病尚無有效的治療手段,以支持治療為主。有文獻報道,輔酶Q、維生素B2可能有效,建議避免極端環境、過量運動、以及服用影響線粒體功能的藥物等[7]。
綜上所述,線粒體神經胃腸型腦肌病因發病罕見,表現復雜多樣,故臨床上極易誤診、漏診。通過本例MNGIE的診治經過及文獻復習希望能加深臨床醫師對MELAS的認識。
