一個2A型肢帶型肌營養不良家系CAPN3基因的突變分析
2020-05-12 02:42:26徐曉薇張新杰王學韜鄒倩倩舒劍波蔡春泉
中風與神經疾病雜志 2020年4期
鄭 潔, 徐曉薇, 張新杰, 王學韜, 鄒倩倩, 舒劍波, 蔡春泉
肢帶型肌營養不良(Limb-girdle muscular dystrophy,LGMD) 是一組以進行性骨盆帶和肩胛帶肌無力為臨床特點的常染色體遺傳性疾病[1]。2018年歐洲神經肌肉疾病中心提出新的命名方式[2],將LGMD分為29種亞型,LGMD D1到LGMD D5為常染色體顯性遺傳;LGMD R1到LGMD R24為常染色體隱性遺傳,這些亞型大多與嚴重先天性肌營養不良相關的基因缺陷有關[3~5]。2A型肢帶型肌營養不良(Limb-girdle muscular dystrophy,LGMD2A)又稱LGMD R1 calpain3-related,是最常見的亞型之一,占LGMD總數的30%,發病率約1/10萬,平均發病年齡17.9歲,由CAPN3基因缺陷導致[6,7]。主要臨床表現為近端肌肉無力、肌萎縮,伴翼狀肩、肌酸激酶升高等。該病臨床表現差異大,臨床表現與X染色體連鎖的杜氏/貝氏進行性肌營養不良相似,易誤診。目前,肌肉鈣蛋白酶-3的缺乏和進一步的CAPN3基因檢測為確診該病的主要方法。在本研究中,使用全外顯子測序方法對1個2A型肢帶型肌營養不良家系進行基因分析以明確病因,為該家系的遺傳咨詢提供依據。
1 對象與方法
1.1 對象 先證者(Ⅱ11),男,現58歲。出生時為足月順產,9歲出現下蹲費力,跑跳困難。19歲出現走路易跌倒,上樓梯需要扶椅,鴨步步態,雙肩抬舉費力。32歲時仍能夠行走,在他人攙扶下可上樓;同年在外院行血液生化檢查:血清肌酸激酶560 U/L(正常參考值50~170 U/L),乳酸脫氫酶98 U/L(正常參考值120~250 U/L),谷草轉氨酶正常;體格檢查:閉眼無力,三角肌、肱二頭肌和肱三頭肌肌力均為3級;髂腰肌、股四頭肌、腓腸肌和脛骨前肌肌力均為2級,腓腸肌無肥大,其余未見異常。肌電圖提示肌源性受損;診斷為肢帶型肌營養不良。46歲時跌倒致一側脛腓骨骨折,后使用輪椅。父母非近親結婚,有家族遺傳史:先證者的二姐(Ⅱ5)和三姐(Ⅱ7)有類似病史,先證者二姐(Ⅱ5)現使用輪椅,可在他人攙扶下行走;先證者三姐(Ⅱ7)現使用輪椅,無法行走。先證者兄長和姐姐均已婚,子女均無發病。先證者父親(Ⅰ1)和長姐(Ⅱ1)已死亡(見圖1)。先證者女兒(Ⅲ7)有再次生育愿望,遂來我院行遺傳咨詢,進行家系基因篩查。本研究獲得家系成員的知情同意,并經醫院倫理委員會批準。
1.2 方 法
1.2.1 樣本 采集收集先證者及其家族成員外周靜脈血2 ml于EDTA抗凝管中,4 ℃保存備用。使用康為世紀生物公司的血液基因組提取試劑盒提取基因組DNA,DNA體積100 μl,濃度達10 ng/μl以上,-20 ℃保存備用。
1.2.2 全外顯子測序 抽取先證者外周血樣1 ml,委托深圳華大基因股份有限公司進行全外顯子組高通量測序。測序原始數據使用BWA軟件與hg19人類參考基因組進行序列比對,采用GATK 軟件進行插入缺失、單核苷酸多態性位點等分析,用Annovar軟件和千人基因組數據庫、dbSNP、OMIM等數據庫進行注釋,利用Polyphen2和SIFT等軟件進行蛋白功能預測。
1.2.3 Sanger測序驗證 根據全外顯子測序結果,采用Sanger測序對先證者及其家系成員進行驗證。PCR擴增家系中其余2例患者和正常家系成員基因。針對CAPN3基因c.1194-9A>G(NM_000070.2)突變設計引物,上游引物為5’-GCCACCCTCTTTTCATCCTCC-3’,下游引物為5’-TGTTCCCACAGTTTCCTGCTTC-3’,Tm值為58 ℃。針對CAPN3基因c.1437C>T(NM_000070.2)突變設計引物,上游引物為5’-TGTAGGGAATAGAAATAAATGG-3’,下游引物為5’-CCAGGAGCTCTGTGGGTCA-3’,Tm值為60 ℃。PCR反應條件為:95 ℃預變性10 min,95 ℃ 30 s,58 ℃/60 ℃ 30 s,72 ℃ 40 s,共35個循環,最后72 ℃延伸10 min。PCR產物使用1.5%瓊脂糖凝膠電泳檢測。使用康為世紀生物公司的瓊脂糖凝膠DNA回收試劑盒純化DNA后,送至北京金唯智生物科技公司進行Sanger測序。測序結果用Chromas軟件與GenBank中參考序列比對,確定突變位點。應用在線軟件Human Splicing Finder(http://www.ummd.be/HSF/)進行基因剪接位點預測。
2 結 果
2.1 全外顯子測序結果 結果顯示先證者CAPN3(NM_000070.2)基因存在復合雜合突變。第9內含子存在c.1194-9A>G雜合突變,生成一個剪切位點突變。在第5外顯子上存在另一個雜合突變,c.1437C>T(p.ser479=),第479位的遺傳密碼子由AGC變成AGT,氨基酸絲氨酸未發生改變的同義突變。檢索ClinVar數據庫,c.1194-9A>G變異為已經報道過的致病性變異;c.1437C>T(p.ser479=)變異未見相關文獻報道,該變異為新突變。
2.2 Sanger測序驗證結果 Sanger測序結果與全外顯子測序結果一致。結果顯示先證者(Ⅱ11)CAPN3基因c.1194-9A>G變異來源于母親(Ⅰ2),c.1437C>T(p.ser479=)未在母親測序結果中發現。家族中患者(Ⅱ5、Ⅱ7)也存在相同的復合雜合突變,其四姐(Ⅱ9)和女兒(Ⅲ7)為c.1194-9A>G變異攜帶者,先證者的女婿(Ⅲ8)未檢測到上述位點變異(見圖2)。
2.3 生物信息學分析 應用在線軟件Human Splicing Finder(http://www.ummd.be/HSF/)評估該變異可能影響剪切,產生新的剪接位點(見圖3)。

A:先證者母親(Ⅰ2)存在c.1194-9A>G雜合變異;B:先證者二姐(Ⅱ5)存在c.1194-9A>G和c.1437C>T的復合雜合突變;C:先證者三姐(Ⅱ7)存在c.1194-9A>G和c.1437C>T的復合雜合突變;D:先證者四姐(Ⅱ9)存在c.1194-9A>G雜合變異;E:先證者(Ⅱ11)存在c.1194-9A>G和c.1437C>T的復合雜合突變;F:先證者女兒(Ⅲ7)存在c.1194-9A>G雜合變異;G:先證者女婿(Ⅲ8)無該位點變異
圖1 先證者家系圖
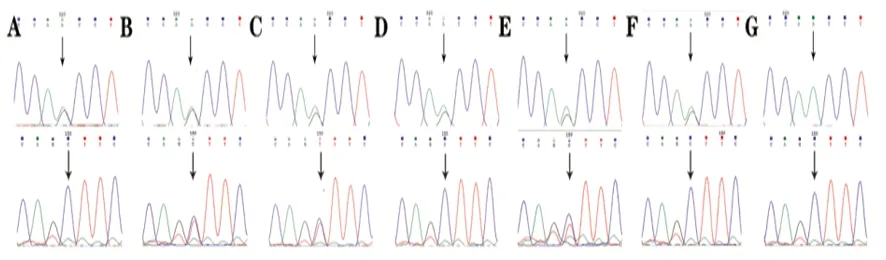
圖2 CAPN3基因兩個突變位點的Sanger測序結果

圖3 生物信息學軟件預測結果圖
3 討 論
LGMDs是一組累及近端肌肉,主要表現為骨盆帶肌和肩胛帶肌無力的常染色體遺傳性疾病。發病率約1/10萬,從2歲至55歲均可發病,平均發病年齡17.9歲。患者自發病至依賴輪椅的平均病程為15.2 y,平均年齡為35.2歲[6]。該病具有高度異質性,肌無力的進展通常是對稱性的,但病情嚴重程度各不相同[8]。LGMD2A的主要臨床特點是進行性肌無力,可表現為跑跳困難、翼狀肩、異常步態等,在病程晚期可有呼吸肌受累導致呼吸衰竭[9,10]。該病的診斷主要依靠臨床表現、肌肉活組織病理學檢查以及基因檢測確定突變,這同時也是進行遺傳咨詢的關鍵[11]。
LGMD遺傳模式分為常染色體隱性遺傳和常染色體顯性遺傳,最常見的LGMD2A型是由CAPN3基因突變引起的常染色體隱性遺傳病[12],目前已經發現了約500個變異,也有報道CAPN3基因內21個堿基對缺失可導致常染色體顯性遺傳[5]。CAPN3基因位于染色體15q15區,該基因由24個外顯子組成,編碼的產物為calpain3[13]。Calpain由821個氨基酸組成,是一種肌肉特異性非溶酶體半胱氨酸蛋白酶,參與肌肉再生、肌膜重構、細胞骨架調控和鈣穩態,與多種關鍵骨骼肌蛋白的分解和裂解相關,尤其是與肌原纖維蛋白骨架構成相關的蛋白,例如肌聯蛋白、細絲蛋白C、黏著斑蛋白等[14~16]。CAPN3基因突變導致這些蛋白活性的喪失,與LGMD2A的發病機制有關。
本研究中,先證者9歲起病,以骨盆帶肌受累為主,出現下蹲費力、跑跳困難,無翼狀肩和腓腸肌肥大,血清肌酸激酶升高,肌電圖提示肌源性受損。先證者于20余年前被診斷為貝氏進行性肌營養不良,但是根據家系調查研究,本家系在Ⅱ代中出現了2名女性患者,后一代男性均無患者,不符合X連鎖隱性遺傳的遺傳模式。全外顯子測序結果顯示先證者CAPN3基因存在一個已知的致病變異c.1194-9A>G,同時檢測到另一同義突變c.1437C>T(p.ser479=),該同義突變在千人基因組及gnomAD數據庫中,東亞人群的頻率為3‰和2.5‰,全人群的頻率為6‰和1‰,不屬于SNP位點。故結合其臨床表現和病史,可確診為LGDM2A。利用Sanger測序對家族成員進行驗證,發現家族中患者(Ⅱ5、Ⅱ7)存在相同的復合雜合突變, c.1194-9A>G變異來源于先證者母親,其四姐(Ⅱ9)和女兒(Ⅲ7)為c.1194-9A>G變異攜帶者。1194-9A>G突變導致新的剪接位點位于正常位點的下游9 bp處,使得第9內含子最后8個堿基得以保留,產生一個新的增強子,這與第7內含子中Alu元件的多次插入有關,使得轉錄本中前7個外顯子缺失[17]。c.1437C>T(p.ser479=)變異在家族中患者(Ⅱ5、Ⅱ7、Ⅱ11)均攜帶,根據美國ACMG遺傳變異分類標準與指南[18],該變異符合致病性證據PM2:ESP數據庫、千人數據庫、EXAC數據庫中正常對照人群中未發現的變異;PM3:在隱性遺傳病中,在反式位置上檢測到致病變異,并已通過患者父母驗證;PP1:突變與疾病在家系中共分離;PP3:預測該變異可能影響剪切;PP4:變異攜帶者的表型或家族史高度符合某種單基因遺傳疾病。符合“2個中等(PM2、PM3)和≥2個支持(PP1、PP3、PP4)”,根據以上證據認為該變異為可能致病的。
綜上所述,本研究中我們對一個LGMD2A型的家系進行基因突變的分析,發現先證者存在CAPN3基因c.1194-9A>G和c.1437C>T(p.ser479=)的復合雜合突變,通過家系研究及生物信息學預測,初步證實了c.1437C>T(p.ser479=)為一導致剪接位點突變的新突變,豐富了CAPN3基因的突變譜,為基因診斷和遺傳咨詢提供了依據。
