以致死性低鉀血癥為首發表現的干燥綜合征1例并文獻復習
2020-03-19 08:16:52鄢巨振
健康研究 2020年1期
孫 忱,鄢巨振
(杭州師范大學附屬醫院 腎病風濕免疫科,浙江 杭州 310015)
干燥綜合征(Sj?gren syndrome, SS)作為臨床最常見的自身免疫性疾病之一,特點為淚腺、唾液腺等外分泌腺體功能受損而致眼干、口干等癥狀[1],主要病理損傷是腺體及內臟組織淋巴細胞浸潤,其腺體外表現不容忽視。本文報道1例罕見的以嚴重低鉀血癥引起周圍性麻痹、呼吸衰竭、心臟驟停為首發且唯一表現的干燥綜合征繼發I型腎小管酸中毒,并復習相關文獻,探討該病的發病機制與治療,以期強調干燥綜合征的腺體外表現可能是首發且致命的,而臨床上不明原因的致死性低鉀血癥需考慮其根本病因為干燥綜合征。
1 病例資料
患者,男,31歲,因“乏力伴肢體活動障礙1天”入杭州師范大學附屬醫院急診室。患者1天前夜間突覺四肢乏力,活動受限,癥狀呈進行性加重,伴呼吸困難,無感覺異常,無意識障礙,無四肢抽搐,無心悸,無惡心嘔吐,無腹瀉等不適,否認外傷史,自述既往無類似病史。患者發病以來,精神差,食欲下降,體重無明顯增減。入院時查體:T 37.6℃,P 65次/分,R 22次/分,BP 119/63 mmHg,神志清,精神軟,皮膚鞏膜無黃染,瞼結膜無蒼白,口唇無紫紺,淺表淋巴結未及腫大,甲狀腺無腫大;雙肺呼吸音清,右下肺可聞及少許濕性啰音,心律齊,各瓣膜區未聞及病理性雜音;腹軟,無壓痛、反跳痛,肝脾肋下未及,移動性濁音陰性,腸鳴音無亢減;雙下肢無水腫,四肢肌力2級,病理征陰性。急診就診期間,患者突發意識不清,呼之不應,口唇發紺,心電圖提示異搏心律,隨之出現心跳驟停,立即予心肺復蘇術,3分鐘后恢復自主心律,心電圖提示竇性自主心律。入重癥監護室后,患者出現呼吸困難,口唇發紺,呼吸球囊輔助呼吸下氧飽和度88%左右,予氣管插管輔助通氣治療。追問患者病史,否認口腔、眼部干燥,否認頭頸部放射治療史,否認利尿劑、糖皮質激素等藥物使用史等。
血氣分析:血液酸堿度7.227,二氧化碳分壓27.00 mmHg,標準碳酸氫根13.2 mmol/L,標準堿剩余15.3 mmol/L;急診電解質:鈉146.4 mmol/L,鉀 1.85 mmol/L,氯121.8 mmol/L;尿常規:尿pH 7.5,尿紅細胞15~20/HP、尿白細胞4~8/HP;自身抗體:抗核抗體陽性(1:320稀釋),抗SSA抗體陽性,抗Ro-52抗體陽性,抗SSB抗體弱陽性;類風濕因子35 IU/mL;血常規:紅細胞132 g/L,白細胞10.34×109/L,血小板221×109/L,生化:肌酐 74.6 μmmol/L,尿素氮4.22 mmol/L,谷丙轉氨酶12 U/L,谷草轉氨酶14 U/L,白蛋白38.4 g/L,球蛋白24.5 g/L,血糖9.4 mmol/L;甲狀腺功能正常;乙肝表面抗原(HBsAg)、丙肝病毒抗體、人類免疫缺陷病毒(HIV)抗體陰性。泌尿系B超:雙腎結石,腎上腺區未見異常包塊回聲;雙側腮腺B超:實質回聲欠均;Schirmer試驗:左3 mm,右5 mm;唇腺活檢:(下唇)小涎腺組織伴間質內多灶淋巴、漿細胞浸潤(淋巴細胞>50個/mm2),見圖1。
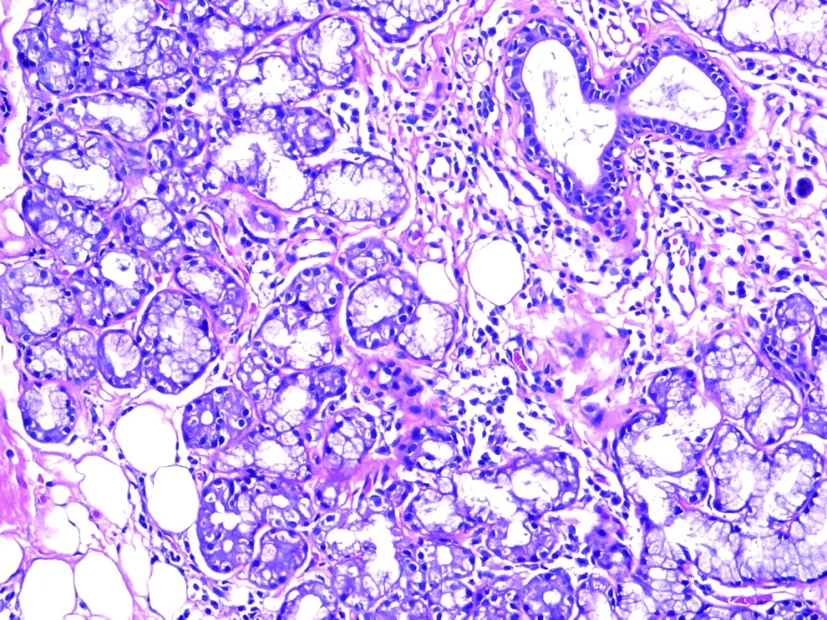
圖1 唇腺活檢(EnVision ×200)
診斷:原發性干燥綜合征,繼發性I型腎小管酸中毒,低鉀血癥,心臟驟停。診斷依據:a.抗Ro/SSA抗體陽性;b.唇腺活檢淋巴細胞灶≥1個/mm2;c.雙眼Schirmer試驗≤5 mm/5min。根據2016年原發性干燥綜合征ACR/EULAR分類標準,評分7分,排除抗膽堿能藥物服用史、頭頸部放射治療史、活動性丙肝、艾滋病、結節病、淀粉樣變、IgG4相關疾病等,無其他結締組織病證據,患者符合原發性干燥綜合征診斷。
治療:予潑尼松30 mg/d治療原發病,予枸櫞酸+枸櫞酸鉀、碳酸氫鈉糾酸補鉀,四肢無力逐漸好轉,呼吸困難、心律失常未再發,5天后血氣、電解質恢復正常,10天后出院。門診隨訪至今,潑尼松規律減量,現潑尼松10 mg/d聯合羥氯喹維持治療。
2 討論
I型腎小管酸中毒又稱遠端腎小管酸中毒(distal renaltubular acidosis,dRTA),發病機制為遠端腎小管上皮細胞泌氫功能障礙,尿液酸化異常,體內H+潴留。其臨床表現為慢性高氯性代謝性酸中毒、低鉀血癥、鈣磷代謝紊亂等,可繼發于自身免疫相關疾病,包括慢性活動性肝炎、系統性紅斑狼瘡、高γ-球蛋白血癥及干燥綜合征等。低鉀血癥是dRTA患者最常見的電解質異常,可導致癱瘓、惡性心律失常、延髓無力和呼吸驟停等,在干燥綜合征患者中曾報道過。
干燥綜合征因瑞典眼科醫生Sj?gren于1933年首先報道而命名,診斷上需與糖尿病、藥物抗膽堿能副反應、頭頸部放療、HIV及丙肝感染、結節病、IgG4相關性疾病、淀粉樣變等多種疾病鑒別。本文以原發性干燥綜合征(primary Sj?gren syndrome, pSS)為主要討論對象。
pSS腎臟累及發生率占4.2%~50%不等[2],以腎間質或小管受累發生率更高,表現為小管間質性腎炎(tubulointerstitial nephritis, TIN),以CD4+T細胞浸潤為主[3]。臨床上,pSS相關TIN表現形式包括遠端小管功能障礙(如遠端腎小管酸中毒、腎性尿崩癥、Gitelman綜合征)、近端小管功能障礙(如Fanconi綜合征)等,可早于口眼干燥癥狀發生[4]。因此,早期識別潛在的pSS腎損害非常關鍵。
dRTA是上述pSS繼發小管損傷中最常見的表現之一,且常隱匿起病,患者無代謝性酸中毒表現,但給予酸負荷下存在尿酸化異常,定義為不完全性dRTA,約占pSS患者的25%,可引起輕度酸血癥及尿pH升高,進而早期出現骨鈣丟失和腎結石[5]。腎小管α-閏細胞上H+-ATP酶和陰離子交換蛋白1(AE1)的低表達被證實與發病相關[6]。針對碳酸酐酶的自身抗體也被報道參與發病[7],部分患者體內可檢測到高水平的抗碳酸酐酶 II、VI和XIII抗體[8]。另一研究認為, S-激酶相關蛋白-1(Skp1)的功能缺陷與之相關,Skp1是參與液泡ATP酶(V-ATPase)組裝調節的組成部分,與pSS特異性抗原Ro-52有一定關聯[9]。抗SSA/Ro抗體和dRTA之間的潛在關聯被也可能是一種致病機制,抗SSA抗體陽性在dRTA患者中較常見[10]。pSS相關dRTA患者通常病程較長,血清β2微球蛋白水平較高,蛋白尿和高血壓的發生率,高于正常腎酸化患者[11]。
目前大多數認為pSS相關dRTA預后相對良好,但由于腎小管萎縮和腎間質纖維化[4],臨床上仍需警惕終末期腎臟病和多器官并發癥發生,建議每6到12個月根據癥狀對pSS患者進行篩查[12]。除對癥治療外,糖皮質激素仍是針對病因的最廣泛治療,各類免疫抑制劑和靶向治療也被應用。但目前仍缺乏大規模前瞻性研究,以評估各類藥物的療效及一線方案。
