非綜合征型唇腭裂核心家庭致病基因突變篩查
2019-11-26 01:53:56茆武熊竹友李薇李光早
山東醫藥 2019年31期
茆武,熊竹友,李薇,李光早
(蚌埠醫學院第一附屬醫院,安徽蚌埠233004)
唇腭裂是口腔頜面部最常見的先天性畸形,據報道,我國新生兒唇腭裂的發病率約為1.82‰[1]。根據全身是否伴有其他畸形,唇腭裂分為綜合征型唇腭裂(SCL/P)和非綜合征型唇腭裂(NSCL/P)。SCL/P的病因較為明確,主要是由染色體異常或單基因突變造成的[2,3],而NSCL/P是一種多基因遺傳病,由遺傳因素和環境因素交互作用引起的[4,5]。NSCL/P具有明顯的遺傳異質性,但基因的篩選、定位及分析方法仍是其病因學研究的難點和熱點。全外顯子組測序技術(WES)是利用序列捕獲技術將全基因組外顯子區域DNA捕捉并富集,然后進行高通量測序的分析方法。WES在篩查范圍和檢出率等方面較其他測序技術具有明顯優勢,如對采用Sanger測序和基因芯片測序不能篩查出基因的樣本,可采用WES進一步篩查鑒定。國內外學者采用連鎖和關聯的分析方法陸續報道了多個與NSCL/P有關的候選易感基因和染色體區域[6,7]。2018年1~6月,本研究通過WES精確定位NSCL/P核心家庭的致病基因,旨在為NSCL/P的產前診斷和預防提供科學依據。
1 對象與方法
1.1 研究對象 選擇同期蚌埠醫學院第一附屬醫院收治的唇腭裂患者,排除范伍德綜合征、歌舞妓面譜綜合征、Meckel綜合征、腭心面綜合征等,診斷為NSCL/P患者6例。所有患者來自不同的核心家庭,父母均為健康人,無先天性疾病。6個核心家庭的基本信息見表1。本研究經蚌埠醫學院第一附屬醫院醫學倫理委員會批準,所有研究對象或其監護人知情同意。

表1 6個核心家庭的基本信息
1.2 基因組DNA提取 采集所有家系成員(患者及其父母)肘靜脈血,采用基因組DNA提取試劑盒提取基因組DNA,經Qubit3.0熒光定量儀鑒定,所提取的DNA濃度和純度合格,可用于后續實驗。
1.3 全外顯子組捕獲和測序
1.3.1 全外顯子組捕獲與建庫 按Agilent Sure Select Human All Exon V6試劑盒說明進行全外顯子組捕獲與建庫。將基因組DNA利用Covaris破碎儀隨機打斷成長度為180~280 bp的片段,經末端修復和加A尾后在片段兩端分別連接上接頭,制備DNA文庫。將pooling后帶有特異index的文庫與生物素標記的探針液相雜交,再用帶鏈霉素的磁珠捕獲基因上的外顯子,PCR擴增后進行文庫質檢,合格即可進行測序。
1.3.2 全外顯子組測序 文庫構建完成后,采用Qubit3.0熒光定量儀定量DNA濃度合格,采用NGS3K/Caliper鑒定文庫的插入片段大小符合預期,采用qPCR法檢測文庫的有效濃度(3 nmol/L),確保文庫質量。文庫質量合格后,運用Illumina測序平臺測序,測序策略PE150,測序深度100X。
1.3.3 單核苷酸多態性(SNP)/插入缺失(InDel)檢測、注釋和篩選 獲取原始測序數據后,在有參考序列或參考基因組(GRCh37/hg19)的情況下,評估測序質量,包括測序錯誤率、數據量和比對率等。測序質量合格后檢測SNP、InDel、拷貝數變異,并統計和注釋檢出的變異情況。
1.4 高級分析流程
1.4.1 基于變異有害性的篩選 ①突變位點篩選:去除在千人基因組數據庫(1000g_all)、ESP6500數據庫(esp6500si_all)、gnomAD數據庫(gnomAD_ALL、gnomAD_EAS)中至少有一個頻率高于1%的突變,得到可能致病的罕見突變;保留處于編碼區或剪接位點區上下10 bp的變異;去除不位于高度保守區且未被軟件預測為會影響剪接的同義SNP突變,或處于重復區的小片段(<10 bp)非移碼InDel突變;保留被預測為有害或影響剪接的突變。②突變位點有害性分類:根據2015年美國醫學遺傳學和基因組學學會(ACMG)制定的變異分類系統[8,9],將突變分為致病的、可能致病的、致病性不明確的、可能良性的、良性的,以此來描述孟德爾疾病致病基因中發現的突變。ACMG的變異分類系統中有28個證據類別,根據這28個證據的組合形式進行變異位點的有害性分類。
1.4.2 新生突變位點篩選 新生突變位點篩選方法:①利用基于家系三成員(患者及其父母)的共同分析,采用SAMtools新生突變位點分析方法,得到患者有而其父母沒有的新生突變位點,經過突變位點篩選得到最終的候選突變位點;②在得到每個家系成員SNP、InDel等突變位點信息的基礎上,篩選患者有而其父母沒有的突變位點作為新生突變位點,經過突變位點篩選得到最終的候選突變位點。參照文獻[10,11]選擇候選突變位點的交集。通過de novo軟件檢測到的de novo位點數計算新生突變速率。
1.4.3 基于候選基因與疾病表型關系的篩選 采用GO功能富集分析和KEGG通路富集分析確定突變基因參與的最主要生化代謝途徑和信號轉導途徑。
1.4.4 蛋白功能互作網絡分析 使用Genemania在線軟件GeneMania對候選基因進行蛋白功能互作網絡分析。
1.4.5 基因-表型-疾病關聯分析 根據提供疾病/表型的名稱,通過精準算法,結合測序結果和多種數據庫(如DisGeNet),對候選基因篩選排序,構建基因-表型-疾病的關聯圖。
1.4.6 候選基因疾病相關性排序 綜合上述基因-表型-疾病關聯分析,針對篩選出的候選基因,依據其與疾病的關聯性強弱進行排序。
2 結果
2.1 患者的新生突變情況 見表2。
2.2 GO功能和KEGG通路富集分析結果 GO功能富集分析能夠生成細胞組件、生物學途徑、分子功能三種結果。其中,富集到生物學途徑的突變基因98個,富集到細胞組件的突變基因56個,富集到分子功能的突變基因97個。KEGG通路富集分析顯示,從PI3K-AKT信號通路中富集到突變基因6個。
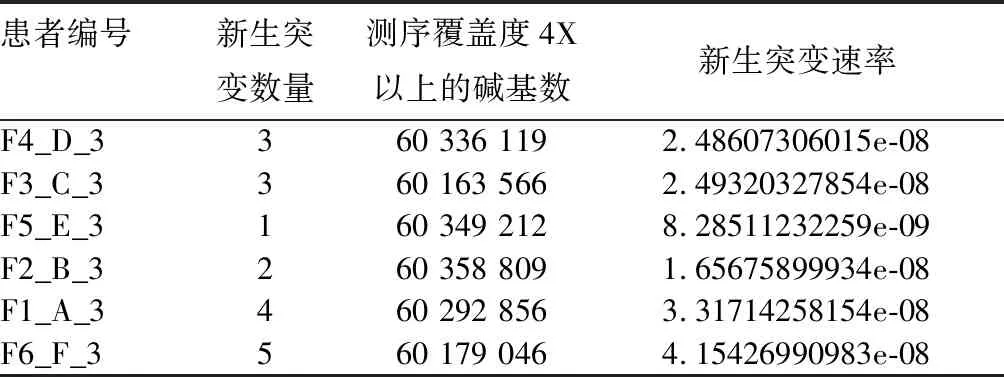
表2 6例患者的新生突變情況
2.3 蛋白功能互作分析結果 見表3。

表3 蛋白功能互作分析結果
注:由于信息量過大,僅取前5行展示。
2.4 候選基因疾病相關性排序 根據篩選出的候選基因,依據其與疾病的關聯性強弱進行排序。排序在前五位的基因分別是PLCG2、DNMT3B、L1CAM、TBL1X、NCOR2。因排序只針對篩選出的候選基因,即便排名在第一位,也不能保證關聯性一定很強。
2.5 候選基因列表 針對篩選出的候選基因,依據ACMG變異分類標準的SNP新生突變位點可能致病性和SNP或InDel的復合雜合標準再進行篩選,共得到10個候選基因、11個突變位點。見表4。
3 討論
NSCL/P是一種多基因遺傳病,其易感基因較多,不同地區人群的易感基因分布特征亦不完全相同。由于受遺傳因素和環境因素交互作用的影響,NSCL/P具有高度遺傳變異性、基因微效性等特征,故NSCL/P易感基因的篩選、定位和分析方法要比SCL/P更加復雜[12~15]。WES是利用序列捕獲技術將全基因組外顯子區域DNA捕捉并富集,然后進行高通量測序的分析方法。在針對孟德爾疾病和復雜疾病的研究中,WES具有明顯優勢。Basha等[16]通過WES對84例NSCL/P患者的易感基因進行篩查,發現了4個罕見的突變基因,分別是TP63、TBX1、LRP6、GRHL3,表明NSCL/P患者可能仍然攜帶與SCL/P相關的突變基因。Liu等[17]通過WES對8例NSCL/P患者的易感基因進行篩查,并通過Sanger測序驗證,共發現了16個與NSCL/P相關的易感基因。這些研究表明,WES在NSCL/P致病基因的篩查方面具有較高的應用價值。
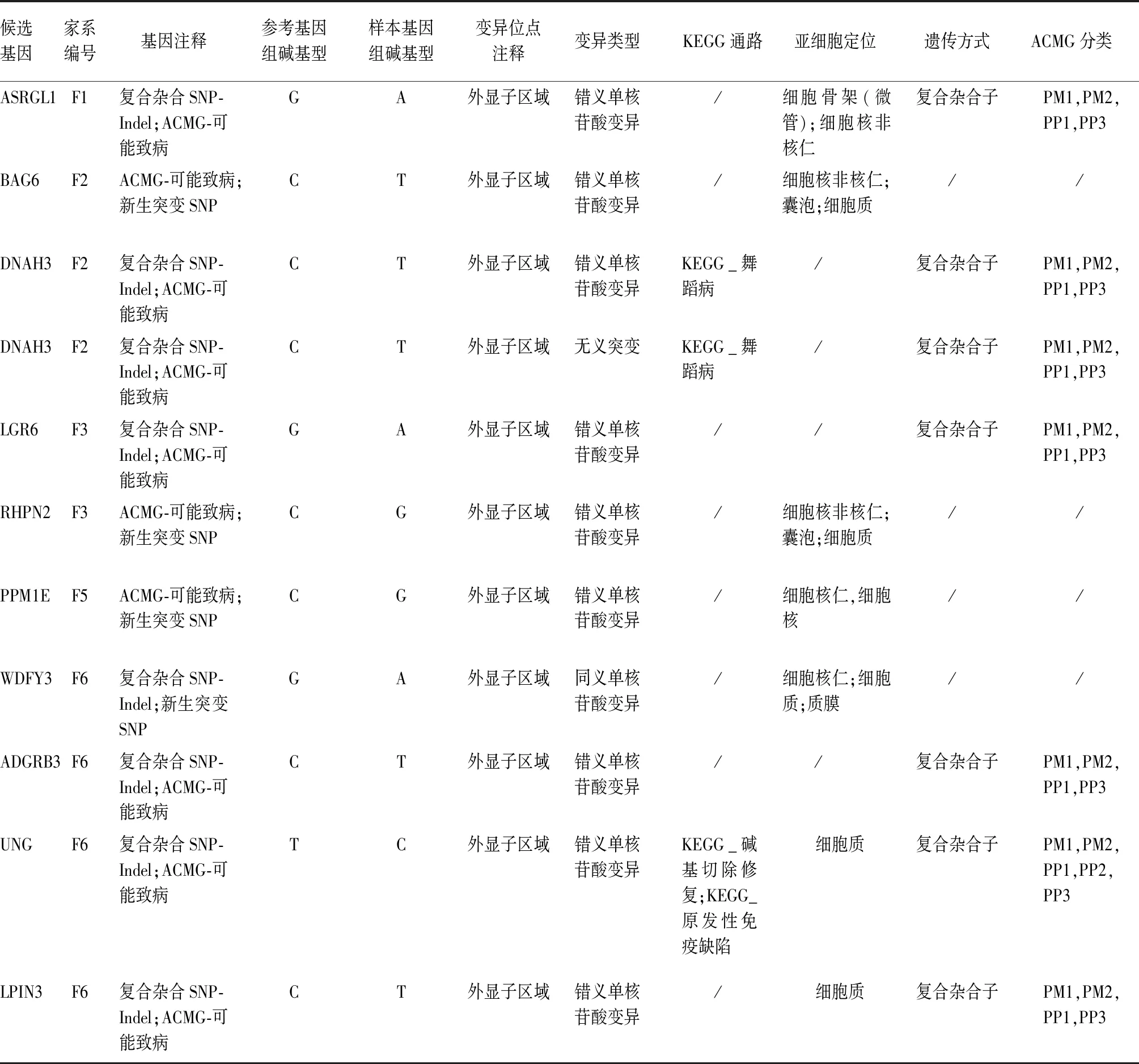
表4 候選基因列表
注:“/”代表未知。
本研究共獲得10個候選基因、11個突變位點,這些突變位點均為SNP位點,有無義突變和終止突變,其中WDFY3的SNP位點是同義突變。按ACMG變異分類標準,BAG6、RHPN2、PPM1E和WDRY3四個基因暫無致病等級,剩余基因均有對應的致病等級,其遺傳模式為復合雜合。在7個有致病等級的基因中,DNAH3基因的KEGG通路富集分析發現與亨廷頓病有關,UNG基因的KEGG通路富集分析發現與堿基切除修復和初級免疫缺陷有關。但本研究未對篩選到的候選基因和SNP位點進行驗證,后續可通過Sanger測序、功能實驗等進一步驗證。
總之,本研究從6個NSCL/P核心家庭中獲得了10個候選基因、11個突變位點,這些候選基因和突變位點可能與NSCL/P發病有關。
