基于碘乙酸功能化樹枝狀聚合物的硫巰化肽段選擇性富集新方法
2019-07-30 09:38:14袁輝明翁葉靖隨志剛張曉丹胡曄晨張麗華張玉奎
色譜 2019年8期
吳 瓊,袁輝明,翁葉靖,隨志剛,張曉丹,胡曄晨,梁 振,張麗華*,張玉奎
(1.中國科學院分離分析化學重點實驗室,中國科學院大連化學物理研究所,遼寧 大連 116023;2.中國科學院大學,北京 100049)
蛋白質的半胱氨酸(Cys)殘基是最具活性的氨基酸殘基之一,不僅經常參與酶反應,而且還能夠形成可逆的氧化翻譯后修飾(oxPTM)[1]。蛋白質的硫巰化修飾,將巰基(-SH)轉換成硫巰基(-SSH),是一種重要的可逆oxPTM[2,3]。作為氣體信號分子H2S信號傳導的一種途徑,自2009年[3]提出以來,蛋白質的硫巰化修飾就引起了廣泛的關注。硫巰化修飾能夠調節蛋白質的活性和功能,如組蛋白脫乙酰酶Sirtuin-1(SIRT1)在Cys371發生的硫巰化修飾,增強SIRT1與鋅離子的結合,進而促進去乙酰化活性[4];三磷酸腺苷合酶α亞基(ATP5A1)在Cys244發生的硫巰化修飾,能夠增強其活性,防止線粒體介導的細胞凋亡[5]。硫巰化修飾的早期研究多數是基于單個蛋白質的鑒定及其后續功能研究[6-8],而組學水平表征硫巰化修飾蛋白質/肽段對其功能的研究具有重要意義,因此利用蛋白質組學的技術手段研究硫巰化修飾必不可少。然而,硫巰化蛋白質組學研究存在很大的挑戰:硫巰基與巰基、二硫鍵相似的化學性質和反應活性使得他們之間難以區分;作為Cys的一種氧化翻譯后修飾,硫巰化修飾肽段的豐度很低[9]。因此,發展高選擇性的富集方法對于硫巰化蛋白質組學的研究必不可少。
隨著蛋白質組學技術的不斷進步,基于硫巰化蛋白質組的富集方法取得了一些重要成果。生物素硫醇法(biotin thiol assay,簡稱BTA)[10]和定量硫巰化位點鑒定法(quantitative persulfide site identification,簡稱qPerS-SID)[11]分別在2015年和2016年提出。BTA和qPerS-SID兩種方法從原理上來說具有一定的相似性:首先,分別利用N-乙基馬來酰亞胺和碘乙酰胺的生物素化試劑封閉蛋白質的巰基和硫巰基,使其生物素化;酶解后,采用鏈霉親和素的瓊脂糖凝珠進行親和富集生物素化肽段;最后,加入還原試劑進行硫巰化肽段的釋放(由-SSH還原成-SH)。此外,BTA方法中引入同位素標記的N-乙基馬來酰亞胺可以實現硫巰化肽段的定量。利用BTA和qPerS-SID方法,Gao等[10]和Longen等[11]分別從小鼠胰腺細胞和HEK293細胞中鑒定到超過1 000和783條硫巰化修飾肽段,有效提高了硫巰化肽段的數據集。在這兩種方法中,由于肽段洗脫時切除了硫巰化修飾位點,有兩種情況可能導致鑒定的假陽性:首先,兩個肽段通過“肽間”二硫鍵相互連接,其中一個肽段包含巰基進而被捕獲,這將導致與硫巰化肽段類似的洗脫行為;其次,基于N-乙基馬來酰亞胺和碘乙酰胺的烷基化試劑對Cys的另外一種氧化翻譯后修飾(亞磺酰化,-SOH)也有微弱的反應活性。因此,BTA方法采取了低濃度的烷基化試劑進行選擇性標記硫巰基,巰基肽段則不與之發生反應,從提高反應特異性角度來降低鑒定假陽性。qPerS-SID方法則引入了基于細胞培養穩定同位素標記技術(stable isotope labeling with amino acids in cell culture,SILAC)進行定量分析來降低鑒定假陽性,并且得出第一種情況的假陽性僅占1.32%,可以忽略不計。這兩種方法均采用生物素化的烷基化試劑標記硫巰化蛋白質,再進行親和富集,由此帶來的缺點是:內源性生物素化肽段的共洗脫會對后續的親和富集造成干擾[12],導致富集選擇性的降低;先標記再富集的多步實驗操作也會造成樣品的損失,不利于低豐度樣品的鑒定。
為了避免內源性生物素化肽段的干擾和簡化實驗流程,本文合成了碘乙酸功能化的聚酰胺-胺樹枝狀聚合物(PAMAM-INS),并結合濾膜輔助的樣品預處理技術用于硫巰化肽段的選擇性富集,簡稱為FADE (filter aided dendrimer enrichment)策略。與BTA和qPerS-SID方法相比,本方法一步實現硫巰化肽段的標記與富集,簡化了實驗流程;不借助生物素與親和素之間的親和富集作用,有效避免了內源性生物素化肽段造成的干擾,有效提高了富集選擇性。為了降低還原洗脫帶來的鑒定假陽性,在FADE策略的基礎上引入SILAC定量分析濃度梯度硫氫化鈉刺激的人神經母瘤細胞(SHSY5Y)中硫巰化蛋白質組,該策略簡稱為增強的FADE策略(eFADE)。
1 實驗部分
1.1 儀器與試劑
采用Spectrum GX紅外光譜儀(鉑金埃爾默,美國)對聚合物進行傅里葉紅外光譜表征;采用全波長酶標儀Multiskan GO(賽默飛科技,美國)進行分光光度測定;采用Ultraflex Ⅲ TOF/TOF質譜儀(布魯克,德國)用于標準肽段和BSA酶解產物的分析;采用EASY-nLCTM1200色譜系統與Orbitrap Fusion Lumos質譜儀(賽默飛科技,美國)構建納升級系統(nano-RPLC-ESI-MS/MS)用于人神經母瘤細胞(SHSY5Y)中硫巰化肽段的富集分析。
截留相對分子質量為10 kDa的濾膜購于賽多利斯(德國);熔融石英毛細管(150 μm i.d.,375 μm o.d.)購自Polymicro Technologies(美國);Venusil XBP C18硅膠填料(粒徑5 μm,孔徑15 nm)購自博納艾杰爾(中國);Reprosil-PurC18-AQ填料(粒徑1.9 μm,孔徑12 nm)購自Dr.Maisch(德國);真空濃縮儀購自賽默飛科技(美國);所有實驗用水均經過Milli-Q(美國)水處理系統處理。
聚酰胺-胺樹枝狀聚合物Generation 6.0(PAMAM,Mr=58 046.11 Da,質量濃度5%)、碘乙酸N-羥基琥珀酰亞胺酯(INS)、牛血清白蛋白(BSA,純度>96%)、牛胰蛋白酶(trypsin)、鹽酸胍(純度≥99%)、蛋白酶抑制劑cocktail、二硫蘇糖醇(DTT,純度≥99%)、碘乙酰胺(IAA,純度≥99%)、5,5′-二硫雙(2-硝基苯甲酸)(DTNB,純度≥98%)、三羥甲基氨基甲烷(Tris,純度≥99.8%)、三(2-羰基乙基)磷鹽酸鹽(TCEP,純度≥98%)、甲酸(FA,純度98%)、硫氫化鈉(NaHS)、三氟乙酸(TFA,純度99%)、氨水(純度≥99.9%)購自Sigma-Aldrich公司(美國);硫化鈉(Na2S,純度>90%)購于百靈威科技有限公司(中國);標準肽段LP9(序列為LEACTFRRP,純度98%)購于強耀生物科技有限公司(中國);乙腈(ACN,純度≥99.9%)、甲醇(純度≥99.9%)、異丙醇(純度≥99.9%)購自Merck公司(德國);α-氰基-4-羥基肉桂酸(CHCA)購于Bruker Daltonics公司(德國);BCA蛋白質濃度測定試劑盒購于碧云天生物科技公司(中國);乙二胺四乙酸(EDTA,純度99%)、氯化鈉(純度≥99.5%)購于科密歐化學試劑有限公司(中國);其他試劑均至少為分析純。
1.2 PAMAM-INS的合成
稱取6 mg INS,加入400 μL甲醇和200 μL,50 mmol/L pH 8.0 PB(磷酸緩沖鹽),振蕩使其完全溶解。稱取32 mg PAMAM溶液,逐滴加入INS溶液中,振蕩使其混合均勻,在室溫進行振蕩反應3 h。反應完畢,將聚合物材料轉移到10 kDa濾膜中,離心去除未反應的INS,用400 μL PB溶液清洗3次,最后加入200 μL PB溶液,得到PAMAM-INS,于4 ℃保存。
1.3 PAMAM-INS中碘乙酰基固載量的測定
采用茚三酮與氨基的顯色法測定聚合物中碘乙酰基固載前后伯氨基含量的差異,以此來計算碘乙酰基的固載量,具體如下:配制0.10、0.20、0.40、0.60、0.80和1.00 mmol/L的正己胺乙醇溶液,利用正己胺做標準曲線,同時做試劑空白對照。
配制0.35%(質量體積分數,下同)的茚三酮乙醇溶液,取100 μL茚三酮乙醇溶液與400 μL上述正己胺系列溶液于60 ℃以1 000 r/min的速度振蕩混勻30 min。反應完畢,取出200 μL紫色反應產物,加到96孔板中,利用全波長酶標儀測定580 nm處的光吸收。以吸光度值為縱坐標、伯氨基濃度(mmol/L)為橫坐標,繪制標準曲線。
配制0.05 g/L PAMAM乙醇溶液和0.05 g/L PAMAM-INS乙醇溶液,分別取出400 μL與100 μL 0.35%茚三酮乙醇溶液反應,反應條件同上(于60 ℃以1 000 r/min的速度振蕩混勻30 min)。反應完畢,進行吸光度值測定。將得到的吸光度值帶入正己胺標準曲線中,算出碘乙酰基固載前后聚合物中伯氨基濃度的差異,即可算出碘乙酰基在聚合物上的固載量。計算公式如下:
(1)
其中,Q為碘乙酰基的固載量(mg/g),C1為PAMAM中伯氨基的濃度(mmol/L),C2為PAMAM-INS中剩余伯氨基的濃度(mmol/L),V為PAMAM-INS的體積(mL),Mr為碘乙酰基的相對分子質量,m為PAMAM-INS的投入量(mg)。
1.4 硫巰化標準肽段的合成
在2 mmol/L標準肽段LP9溶液中,加入20 mmol/L的DTNB溶液,于37 ℃反應30 min后,再加入200 mmol/L的Na2S溶液,于37 ℃反應30 min后進行液相色譜的除鹽,凍干。復溶于水,使用完畢通氮氣進行保存,防止硫巰化修飾發生氧化。
1.5 標準蛋白BSA的酶解
將1 g/L BSA溶液于95 ℃變性5 min,加入終濃度為10 mmol/L的DTT,于56 ℃還原1 h。隨即加入終濃度為25 mmol/L的IAA進行烷基化30 min,加入25 μg trypsin,于37 ℃水浴中酶解10 h。加入甲酸終止反應,進行液相色譜的除鹽。
1.6 FADE策略的抗干擾能力評價
將合成的硫巰化標準肽段LP9與BSA的酶解產物按照質量比1∶100混合,隨即轉移到合成好的PAMAM-INS中,于40 ℃在10 kDa濾膜中反應12 h。反應完畢,分別采用2 mol/L氯化鈉溶液、4%氨水溶液、50%異丙醇水溶液(含0.1%TFA)、蒸餾水清洗濾膜各兩次。于56 ℃水浴中,加入終濃度為10 mmol/L的TCEP進行硫巰化肽段的釋放,離心得到洗脫液。將標準肽段LP9與BSA的酶解產物的混合物(富集前樣品)、最后一次的非特異性吸附淋洗液以及富集洗脫液分別進行MALDI-TOF檢測。
1.7 SHSY5Y細胞的培養和蛋白質提取
SHSY5Y細胞培養:分別用天然同位素氨基酸(12C6-lysine和12C614N4-argine)和重同位素標記的氨基酸(13C6-lysine和13C615N4-argine)、10% FBS以及1% DMEM培養基置于37 ℃恒溫培養箱中,在5% CO2氣體濃度條件下進行培養細胞,經過多次傳代使細胞標記完全,得到SILAC輕標(12C6-lysine和12C614N4-argine)和重標(13C6-lysine和13C615N4-argine)的SHSY5Y細胞,其中重標細胞平行培養3份。待細胞長到80%~90%的融合度之后,在3份重標細胞中分別加入終濃度為30、60和100 μmol/L的硫氫化鈉溶液刺激細胞30 min。收集細胞懸液,反復吹打,以1 551 r/min的速度離心收集細胞,然后用1×PBS(磷酸緩沖鹽,pH 7.4)清洗細胞3次。
分別提取4份細胞(1份輕標、3份重標)的蛋白質:將細胞分散在6 mol/L鹽酸胍中,按照1%的體積比加入蛋白酶抑制劑cocktail,在冰浴中進行超聲破碎。最后以14 725 r/min的速度離心40 min,收集上清,采用BCA法測定蛋白質濃度。
1.8 eFADE策略用于SHSY5Y細胞中硫巰化肽段的選擇性富集
將輕標的SHSY5Y蛋白質溶液和3份重標的SHSY5Y蛋白質溶液分別按照各自的蛋白質濃度進行等質量的混合,得到3份輕重標混合的樣品(每份均為1 mg蛋白質)。在95 ℃下變性10 min,隨即轉移到10 kDa濾膜中,離心去除鹽酸胍。在每個濾膜中加入100 μL PB溶液,按照酶與蛋白質的質量比為1∶25加入胰蛋白酶,將濾膜置于37 ℃水浴中酶解,酶解溶液進行通氮氣保護。16 h后,將濾膜進行離心,得到酶解產物。將3份酶解產物分別轉移到等質量的PAMAM-INS中(保存在濾膜中),避光室溫混勻12 h,分別進行3份樣品的硫巰化修飾肽段的富集。分別采用2 mol/L氯化鈉溶液、4%氨水溶液、50%異丙醇水溶液(含0.1%TFA)、蒸餾水清洗濾膜各兩次,洗去非特異性結合的肽段。在56 ℃水浴中,每個濾膜加入終濃度為10 mmol/L的TCEP進行硫巰化肽段的釋放,離心得到洗脫液。在洗脫液中,加入終濃度為20 mmol/L的IAA進行烷基化,除鹽后凍干,進行Orbitrap Fusion Lumos質譜分析。
1.9 MALDI-TOF MS分析
將1 μL待分析物與1 μL CHCA基質(7 g/L溶于含0.1%TFA的60% ACN溶液)依次點于MALDI靶板上。采用固體激光Smart Beam技術(355 nm)及正離子反射模式進行MALDI-TOF MS分析。
1.10 Nano-RPLC-ESI-MS/MS分析及數據處理
采用EASY-nLCTM1200色譜系統與Orbitrap Fusion Lumos質譜儀構建納升級系統用于SHSY5Y細胞中硫巰化肽段的鑒定。Nano-RPLC的分離條件如下:C18毛細管預柱(3 cm×150 μm,Venusil XBP C18,粒徑5 μm,孔徑15 nm)和C18毛細管分離柱(15 cm×150 μm,Reprosil-PurC18-AQ,粒徑1.9 μm,孔徑12 nm);流動相A:0.1%FA;流動相B:80%ACN+0.1%FA;流速:600 nL/min;流動相梯度設置:0~55.0 min,10.0%B~28.0%B;55.0~70.0 min,28.0%B~47.0%B;70.0~72.0 min,47.0%B~95.0%B;72.0~75.0 min,95.0%B。
Orbitrap Fusion Lumos質譜參數如下:質譜采集在數據依賴型(DDA)正離子模式下進行,完成一次全掃描及二級掃描的時間為3 s;噴霧電壓為2.1 kV;離子傳輸毛細管溫度為320 ℃;MS的掃描范圍為m/z350~1 500,分辨率設為60 000,自動增益控制(AGC)設為4×105,離子最大累積時間設為50 ms;母離子采用高能碰撞碎裂模式(HCD)進行二級碎裂,碎裂能量為34%;MS/MS的分辨率設為15 000,AGC設為5×104,離子最大累積時間設為35 ms;動態排除窗口時間為20 s;選擇電荷價態+2~+7的肽段進行碎裂。
將Orbitrap Fusion Lumos質譜儀上產出的raw文件首先用Proteome Discoverer軟件(v2.2)中整合的Mascot(v2.4)進行數據庫檢索。FASTA數據庫為UniProtKB human(release 2018_1119)。一級母離子的質量容忍度設置為10 ppm(10×10-6);二級碎片離子的質量容忍度設置為20 mmu。FASTA數據庫在搜索前會被搜庫軟件自動轉換成正-反庫的形式。檢索設置參數包括:甲硫氨酸的氧化(+15.99 Da)和蛋白質的N端乙酰化(+42.01 Da)設為可變修飾;添加SILAC標記賴氨酸(K6,+6.02 Da)和精氨酸(R10,+10.00 Da)及半胱氨酸的烷基化(+57.02 Da)設為固定修飾;添加胰蛋白酶的特異性酶切,并允許最多2個漏切位點。蛋白質鑒定至少包含一個唯一性肽段;輸出結果中H/L定義為重標與輕標的比值。檢索結果采用Proteome Discoverer軟件中的Percolator模塊進行過濾:控制肽段的FDR不大于1%,其他設置均為軟件默認。
1.11 生物信息學分析
利用在線工具PANTHER(v14.0,http://pantherdb.org/)對硫巰化蛋白質參與的生物學過程、分子功能和通路進行分析。氨基酸序列分析采用pLogo(v1.2.0,https://plogo.uconn.edu/)。
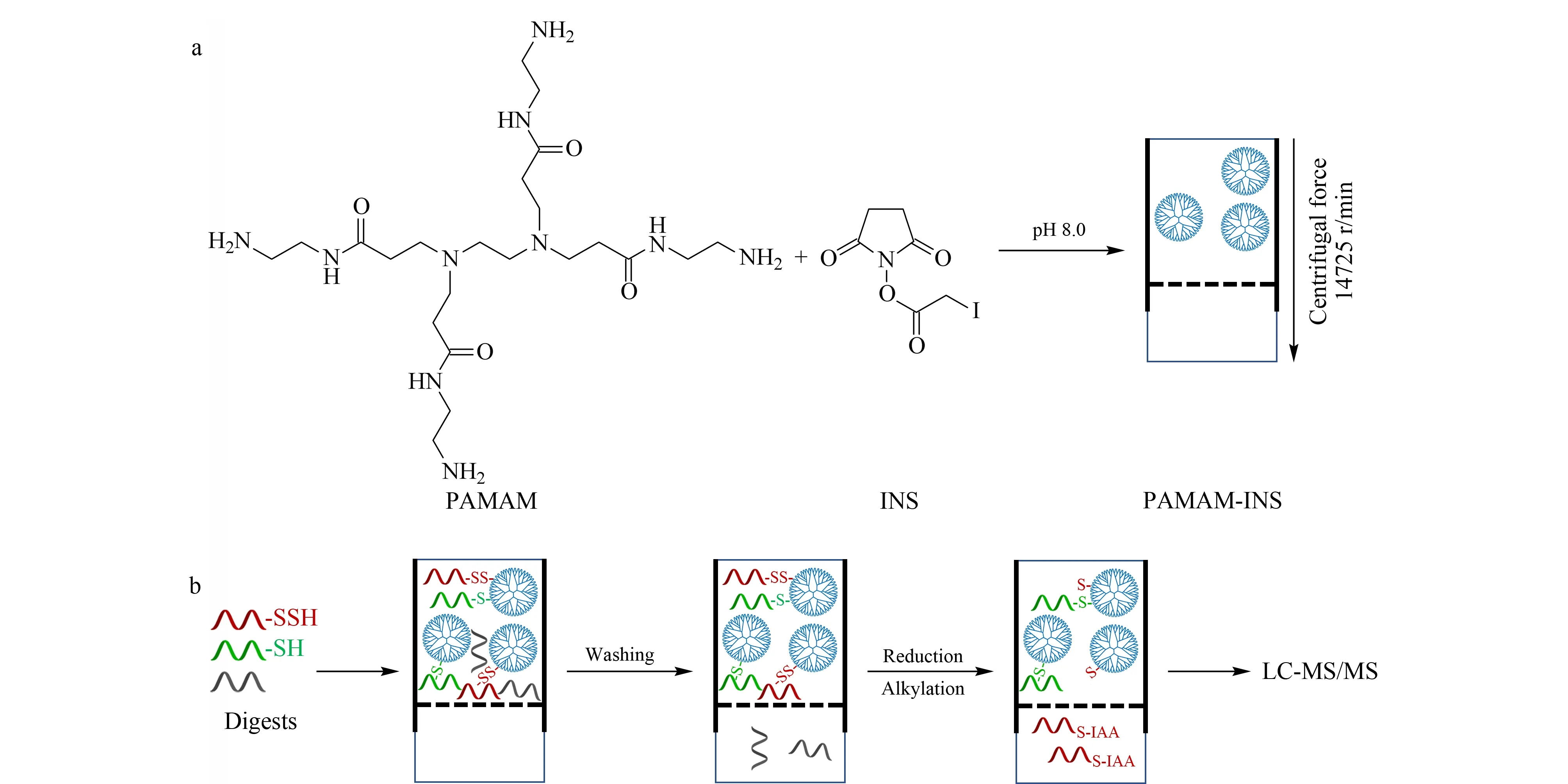
圖1 (a)PAMAM-INS的制備及(b)FADE策略用于硫巰化肽段的富集流程Fig.1 Workflow of(a)the preparation of PAMAM-INS(polyamidoamine-iodoacetic acid N-hydroxysuccinimide ester)and (b)the FADE(filter aided dendrimer enrichment)strategy for the enrichment of persulfidated peptidesIAA:iodoacetamide.
2 結果與討論
2.1 PAMAM-INS的制備及FADE策略的建立
PAMAM是一類具有高度支化三維結構的大分子[13],第六代的PAMAM外層含有256個表面伯氨基(58 046.11 Da),具有很好的生物相容性和親水性。碘乙酸N-羥基琥珀酰亞胺酯(INS)是一種雙異官能團交聯劑,一端帶有的N-羥基琥珀酰亞胺酯可以偶聯到PAMAM表面的伯胺上,另一端的碘乙酰基可以與蛋白質的巰基/硫巰基進行特異性次級鍵合。基于上述化學反應,通過一步法合成了具有巰基和硫巰基反應活性的PAMAM-INS。如圖1a所示,在避光的堿性條件下(pH 8.0),將INS溶液與PAMAM溶液進行均相反應(其中,INS物質的量相較于PAMAM是過量的)后轉移至10 kDa濾膜,反應產物PAMAM-INS則被截留在濾膜上,通過離心可去除過量的INS。該制備策略具有如下優點:操作簡單(一步即可合成)、液-液的均相反應體系加快傳質過程,整個制備過程耗時不到5 h。因此,結合PAMAM-INS聚合物溶液的特性與濾膜輔助的樣品預處理,FADE策略可以實現硫巰化肽段的一步標記與富集,簡化了實驗操作。具體過程如圖1b所示,將實際樣品的蛋白質酶解產物(不進行還原和烷基化處理)轉移至濾膜中與PAMAM-INS聚合物溶液進行孵育,巰基和硫巰基肽段則被PAMAM-INS捕獲從而截留在濾膜上,離心洗除非特異吸附之后,加入還原劑進行硫巰化肽段的洗脫(而巰基肽段仍被截留在濾膜上),烷基化后進行質譜鑒定,從而實現硫巰化肽段的選擇性富集。
2.2 PAMAM-INS的紅外光譜表征
對反應物及其產物進行了紅外光譜的表征,如圖2所示,在反應物INS中,1 724 cm-1屬于酮基C=O的伸縮振動峰,1 044 cm-1屬于C-N的伸縮振動峰,545 cm-1應屬于C-I的伸縮振動峰。在PAMAM中,3 285 cm-1屬于伯胺N-H的伸縮振動峰,2 938 cm-1和2 820 cm-1屬于亞甲基的伸縮振動峰,1 633 cm-1屬于酰胺鍵中C=O的伸縮振動峰,1 541 cm-1屬于N-H的彎曲振動峰,1 026 cm-1屬于C-N的伸縮振動峰。PAMAM-INS與PAMAM的譜峰類似,但是在600~500 cm-1區間內多了533 cm-1的吸收峰,歸屬于C-I的伸縮振動峰,證明PAMAM-INS的制備成功。
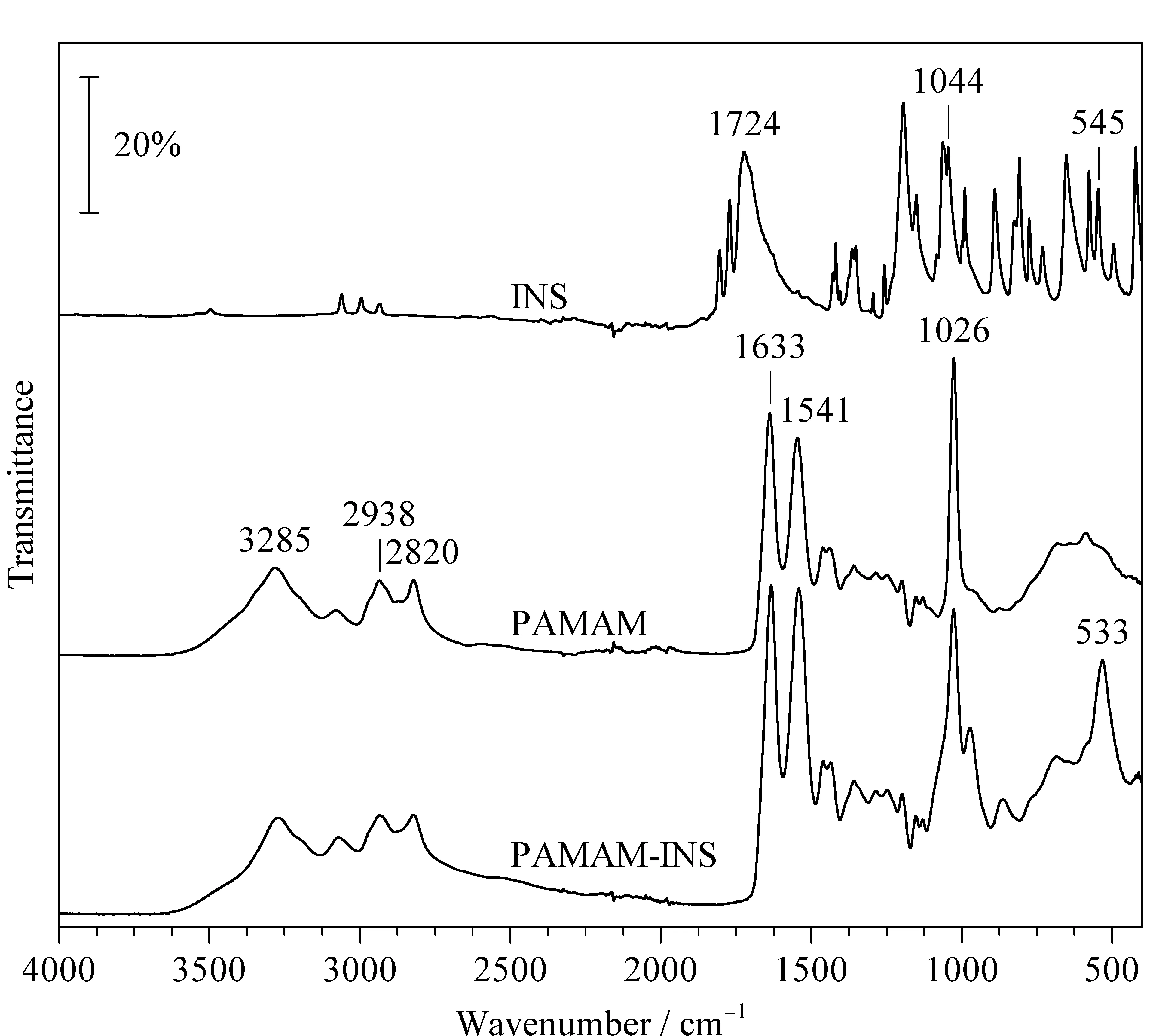
圖2 INS、PAMAM及PAMAM-INS的傅里葉紅外光譜圖Fig.2 FT-IR spectra of INS,PAMAM,and PAMAM-INS
2.3 PAMAM-INS中碘乙酰基固載量的測定
采用茚三酮與氨基顯色法測定聚合物中碘乙酰基固載前后氨基含量的差異,從而計算得到碘乙酰基的固載量,其顯色原理為伯氨基與茚三酮共熱生成NH3和還原性茚三酮,進一步縮合成藍紫色的茚二酮胺。該化合物顏色的深淺與伯氨基含量成正比,可通過測定580 nm處的光吸收計算碘乙酰基固載前后聚合物中伯氨基含量的差異,通過伯氨基含量的減少計算得到碘乙酰基的固載量(在PAMAM與INS反應中,PAMAM的伯氨基物質的量減少量即為碘乙酰基的鍵合物質的量)。通過帶入正己胺標準曲線中(其線性回歸方程為y=0.344 3x-0.001 3,y為吸光度值,x為伯氨基的濃度,單位為mmol/L;相關系數R2=0.99),測得反應前后伯氨基濃度分別為0.92和0.40 mmol/L,即鍵合的碘乙酰基濃度為0.52 mmol/L,從而計算出碘乙酰基在PAMAM-INS中的固載量為2.20 g/g。此外,樹枝狀聚合物PAMAM-INS中超過一半的氨基被碘乙酰基取代,高的碘乙酰基鍵合量有利于提高硫巰化肽段的結合容量。
2.4 FADE策略抗干擾能力的評價
在文獻[14]的基礎上略加修改合成了硫巰化標準肽段LP9(序列為LEACTFRRP),將合成的硫巰化標準肽段摻到100倍質量干擾的BSA酶解產物中,對FADE策略的抗干擾能力進行考查,結果采用MALDI-TOF檢測。如圖3a所示,富集前由于BSA肽段的信號抑制,硫巰化標肽無法檢測到。富集過程中,采用嚴苛的條件進行非特異吸附的清洗,并且在最后一次清洗非特異吸附過程中,檢測不到任何肽段(見圖3b)。
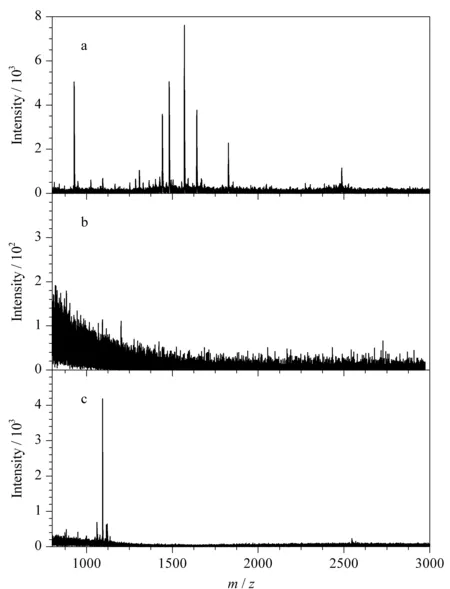
圖3 硫巰化標肽和BSA酶解產物質量比為1∶100的混合溶液的MALDI-TOF質譜圖Fig.3 MALDI-TOF mass spectra of persulfidatedstandard peptides and BSA digests mixtures at1∶100 (m/m) a.before enrichment;b.the last washing;c.after the enrichment by FADE strategy.
加入還原劑進行洗脫后,可以看到硫巰化標肽切除之后形成的巰基肽段,且其信號得到很大幅度的提高(見圖3c)。由此說明FADE策略對硫巰化肽段有很好的富集效果,有望進一步應用到復雜樣品中硫巰化肽段的富集。
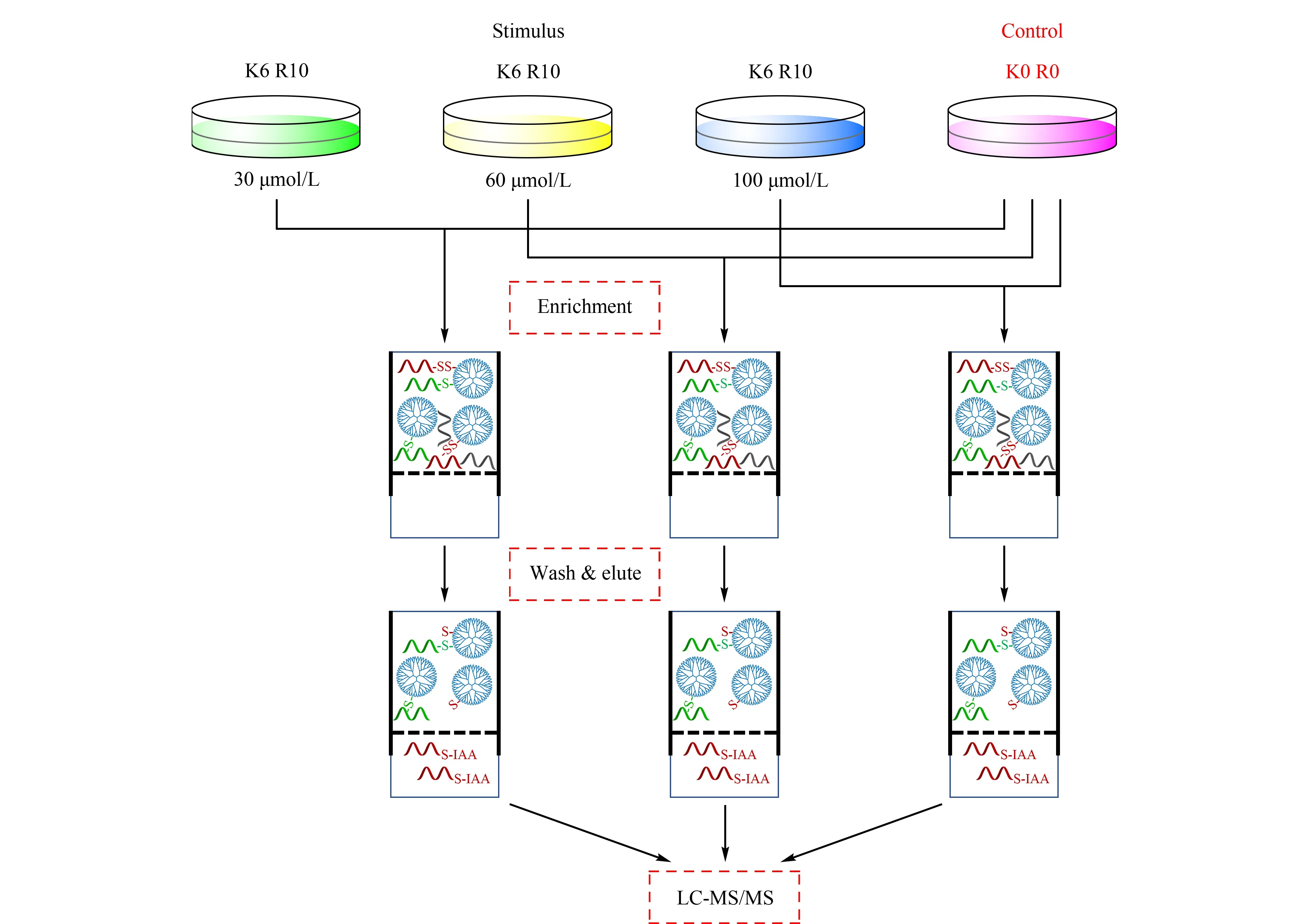
圖4 eFADE策略用于SHSY5Y細胞中硫巰化肽段的富集Fig.4 Application of the eFADE(enhanced filter aided dendrimer enrichment)strategy for the enrichmentof persulfidated peptides from SHSY5Y cells K0R0 and K6R10 are referred to as light and heavy cultured cells,respectively.
2.5 eFADE策略用于實際樣品中硫巰化肽段的鑒定

圖5 SHSY5Y細胞中硫巰化肽段的鑒定結果Fig.5 Identification results of persulfidated peptides from SHSY5Y cells a.the number of identified thiol-containing peptides in the eluant;b.the box plot of the quantification ratios of the thiol-containing peptides in the eluant;c.the quantified thiol-containing peptides exhibiting a positive trend.The heavy to light ratio of cysteine peptides in the eluant was identified as H/L.
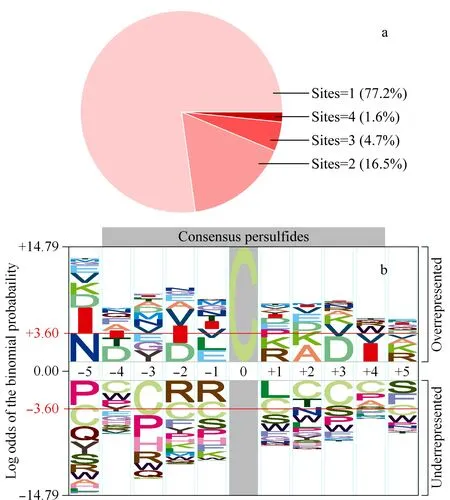
圖6 (a)每個蛋白質中硫巰化位點的分布及(b)pLogo分析硫巰化位點的保守氨基酸序列Fig.6 (a)Distribution of the number of persulfidatedsites per protein,and(b)the consensus sequence motifs of persulfidated cysteines visualized by pLogo
在FADE策略中,由于洗脫時切除了硫巰化修飾位點,可能導致鑒定的假陽性,因此建立了eFADE策略。eFADE策略具體思路如圖4所示,利用SILAC技術對細胞進行代謝標記,并在重標標記的細胞中加入不同濃度的NaHS(H2S供體)刺激30 min,產生的H2S能誘導細胞內硫巰化修飾水平的增加[15,16]。通過設置濃度梯度刺激細胞,預期使細胞內硫巰化修飾水平發生不同幅度的增加。在eFADE策略中洗脫得到的巰基肽段中都有一個定量比值(重標/輕標),選取3組刺激條件中共同鑒定到且定量比值隨著NaHS刺激濃度增加呈上升趨勢的巰基肽段作為硫巰化肽段,以此來降低鑒定的假陽性。利用eFADE策略,在30、60和100 μmol/L硫氫化鈉刺激的SHSY5Y細胞中,分別鑒定到624、587和821條巰基肽段(見圖5a)。該方法對硫巰化肽段的平均富集選擇性為77.20%,高于qPerS-SID方法報道的富集選擇性(56%)[11],說明該均相富集過程有利于提高富集選擇性。如圖5b所示,在鑒定到的巰基肽段中,其定量比值的中位數隨NaHS濃度增加呈上升趨勢,這說明隨著NaHS刺激濃度的增加巰基肽段有上調趨勢,暗示硫巰化修飾發生上調,與預期相符合,即采用濃度梯度的NaHS刺激細胞能使細胞內硫巰化修飾水平發生不同幅度的增加。因此,按照3組刺激條件中共同鑒定到且定量比值隨著NaHS刺激濃度增加呈上升趨勢的標準進行篩選,得到163條硫巰化肽段,對應127個蛋白質(見圖5c)。其中,發現了一些已被驗證過的硫巰化修飾蛋白質,如磷酸甘油酸脫氫酶(PHGDH)、丙酮酸激酶(PKM)、肌動蛋白(ACTB)等[3,10,11]。與文獻報道[11]的結果相比,34.70%的硫巰化肽段與qPerS-SID報道的結果相同,表明兩種方法具有一定的互補性。在SHSY5Y細胞中,E3泛素連接酶parkin的硫巰化修飾有助于帕金森疾病的治療[17]。雖然在本試驗中沒有鑒定到硫巰化的parkin蛋白質,但鑒定到了其他與帕金森疾病相關的硫巰化蛋白質,如類泛素修飾激活酶1(UBA1)、E2泛素連接酶(UBE2R2)、增殖細胞核抗原(PCNA)等,暗示硫巰化修飾在帕金森這種神經退行性疾病中可能扮演重要角色。對鑒定到的蛋白質含有的硫巰化位點進行了統計,如圖6a所示,77.20%的蛋白質只含有一個硫巰化修飾位點;超過6%的蛋白質包含3個及以上的硫巰化位點,表明這些蛋白質發生了廣泛的硫巰化修飾并且對H2S比較敏感。分析硫巰化修飾位點附近的氨基酸分布情況,有助于預測潛在與硫巰化修飾類型相關的保守序列區間。采用pLogo對硫巰化修飾位點進行motif分析,如圖6b所示,修飾位點附近存在較明顯的帶負電氨基酸(天冬氨酸,D),與文獻[11]報道的結果類似,帶負電的氨基酸有利于穩定硫巰化位點。
2.6 生物信息學分析
為了進一步研究硫巰化蛋白質的功能,利用在線的生物信息學工具PANTHER對硫巰化蛋白質進行了GO和通路的分析(見圖7)。生物學過程的分析結果顯示,硫巰化蛋白質參與了生命活動的多個過程,如核糖體蛋白復合物的自組裝、蛋白復合物亞基的組織、形成翻譯起始三元復合物等,還包括多個與翻譯相關的過程(翻譯起始、翻譯終止、細胞質翻譯)。分子功能的分析結果顯示,硫巰化蛋白質發揮結構分子活性、翻譯起始因子活性以及各種結合功能等。PANTHER通路的分析結果顯示,硫巰化蛋白質參與糖酵解、亨廷頓疾病、Slit/Robo介導的軸突導向、整合素信號通路等信號通路中。其中,糖酵解和Slit/Robo介導的軸突導向通路的富集效果最明顯,富集倍數分別是30.50和20.34。據文獻[10]報道,硫巰化修飾可以調節糖酵解通量,引起細胞能量的重新編程和代謝,本結果也顯示硫巰化修飾與糖酵解信號通路密切相關。此外,神經導向因子Slit家族是一類分泌型糖蛋白,可通過結合神經細胞膜上的Robo受體家族發揮生物學功能。Slit/Robo介導的神經軸突導向參與調控發育中的中樞神經軸突定向延伸,促進軸突生長和調控神經纖維束化、定位以及突觸的形成[18,19]。由此,猜測硫巰化修飾可能在中樞神經系統扮演重要角色。
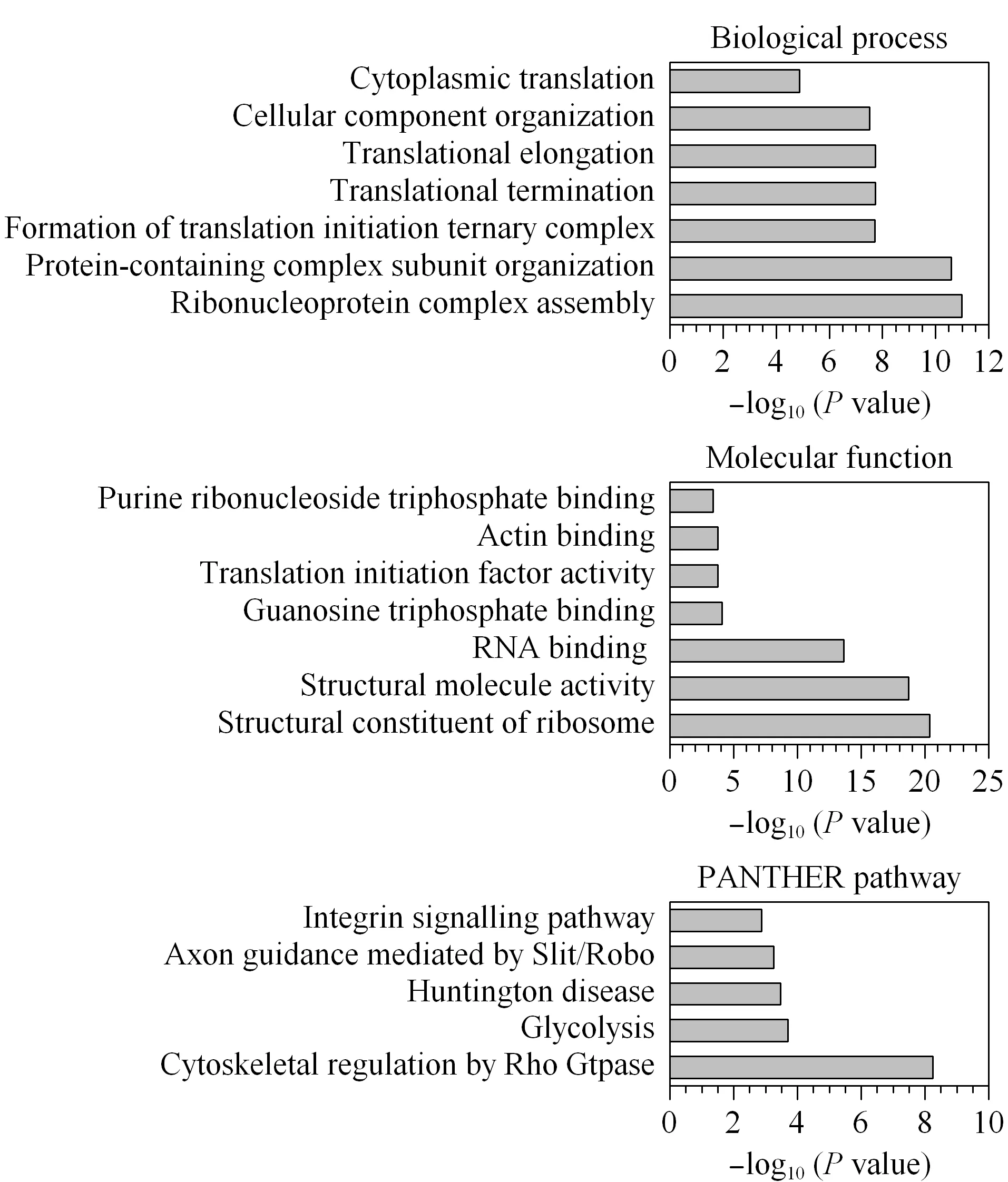
圖7 使用PANTHER進行GO富集分析生物學過程、分子功能和通路分析Fig.7 GO term enrichment analysis of biologicalprocesses,molecular function,and pathway using PANTHERGTP:guanosine triphosphate.
3 結論
本文合成了PAMAM-INS用于硫巰化肽段的選擇性富集,進一步結合SILAC技術與濃度梯度刺激細胞進行定量分析來降低鑒定的假陽性。與文獻[10,11]報道的方法相比,該方法一步實現了硫巰化肽段的標記與富集,簡化了實驗流程;不借助生物素與親和素之間的親和富集,避免了內源性生物素化肽段造成的干擾,有效提高了富集選擇性。將eFADE策略應用于SHSY5Y細胞中硫巰化肽段的富集,生物信息學的結果暗示硫巰化修飾可能在中樞神經系統中扮演重要角色。這些結果表明,本文發展的eFADE策略將為規模化分析硫巰化蛋白質組學提供重要的研究工具。
