基于R語言爬蟲對Illumina接頭序列的挖掘
2019-06-10 09:31:49柏程思
科技創新導報 2019年5期
柏程思
摘 ? 要:當前生物信息學過濾測序接頭序列的軟件不能涵蓋所有Illumina測序平臺的接頭序列。這樣造成了分析NGS數據平臺的局限性。本文通過R語言編程利用爬蟲技術對Illumina質控文件的分析,挖掘所有不能被過濾軟件識別的接頭序列。
關鍵詞:生物信息學 ?R語言編程 ?爬蟲 ?Illumina測序
中圖分類號:Q811.4 ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ?文獻標識碼:A ? ? ? ? ? ? ? ? ? ? ? 文章編號:1674-098X(2019)02(b)-0136-02
當前生物信息測序領域中,Illumina公司屬于二代測序的壟斷公司。其開發的Illumina Hiseq、Illumina Miseq、Illumina GAII等平臺已經是流行于全世界。絕大多數分子生物學、基因組學和細胞生物學實驗室都在使用Illumina平臺測序。
在二代測序的分析流程中,拿到的數據是FASTQ數據,需要先對數據進行質量控制。質量控制通常是使用FastQC軟件對FASTQ數據進行分析,判斷測序數據是否具有高質量。如果質量低,則不支持后續生物信息學分析,需要過濾。通常情況下,由于測序儀機器的誤差,從測序儀下機的數據都或多或少有低質量的序列,這些低質量需要有的是堿基質量低,有的是測序接頭未去掉(盡管Illumina大多數測序平臺的測序儀在2013年以后能保證數據下機自動去接頭,但是部分測序平臺依然不能自動去接頭)。需要過濾,而過濾使用的軟件一般為Trimmomatic軟件。但是Trimmomatic軟件過濾使用的文件是自身adapter文件夾中自帶的Truseq文件過濾測序的接頭,而這些接頭文件只包含了Hiseq、Miseq和GAII測序平臺的接頭文件,沒有包括全部的接頭文件。未去接頭的序列在質控結果中可以將接頭序列以Overrepresent形式表示出來。所以,如果我們測序時選擇的測序平臺不能自動下機去接頭,我們需要手動自己去接頭。爬蟲是利用計算機技術對網絡數據的挖掘,因為互聯網數據基本都是儲存在網絡服務器中,網絡服務器末端端口是用戶。用戶可以通過網頁訪問網絡服務器,網頁是由HTML語言搭建的可視化端口。HTML是HyperText Markup Language(超文本標記語言)的縮寫,這個語言使用<標簽>內容基本格式進行網頁編輯[1]。例如
This is how to use HTMLThe way to use HTML
You can learn it
將上面這個代碼復制到一個新建TXT文本中,并將后綴命名為.html,雙擊該文件打開會出現以下內容(見圖1)。
1 ?分析方法
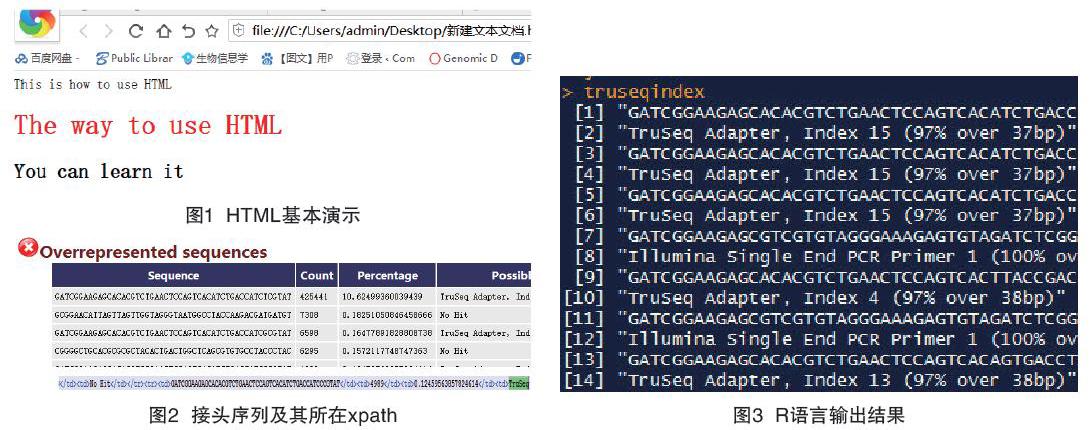
FASTQC軟件輸出的質控結果就有HTML本地文件,如果出現了接頭序列就會在Overrepresent中出現,Overrepresent有其對應的HTML標簽。R語言[2-3]可以通過追溯內容所在的標簽追溯到內容,這個追溯內容的路徑稱之為xpath,最后通過正則表達式篩選我們要的內容即可。首先在Linux系統上存放測序數據的路徑(該路徑中只能含有測序數據文件)下使用命令fastqc `ls $pwd`,然后下載輸出的HTML文件。先用網頁查看是否有接頭序列,再用Notepad++打開文件找接頭序列所在的xpath(見圖2)。
編寫如下R語言代碼
library(rvest)
library(stringi)
setwd("D:/test/fastQC")
myQCfile<-dir("D:/test/fastQC")
truseqindex<-NULL
for (i in 1:length(myQCfile)) {
qc<-read_html(myQCfile[i])
a<-qc%>%
html_nodes(xpath = "http://tr/td")%>%
html_text()
b<-NULL
for (j in 1:length(a)) {
if(grepl(a[j],pattern = "(TruSeq|Primer)")){b<-c(b,a[j-3],a[j])} }
truseqindex<-c(truseqindex,b)}
2 ?結語
我們開發的挖掘當前過濾軟件無法過濾的接頭腳本更具有實用性,可以適用于所有NGS數據分析過濾腳本。使分析結果更具有可靠性。
參考文獻
[1] 鄧子云.爬蟲系統中標簽刪除功能的設計及優化[J].計算機系統應用,2019,28(1):176-181.
[2] 許慶煒.B語言—生物信息學可視化流程語言[J].計算機與數字工程,2009,37(5):90-93.
[3] 吳棟楊. 構建基于R語言的生物信息學研究平臺[A].第十次中國生物物理學術大會論文摘要集[C].中國生物物理學會,2006:1.
