頭孢拉定合成工藝的優化
2019-03-13 08:40:44李文杰曹桂僖
國外醫藥(抗生素分冊) 2019年1期
李文杰,曹桂僖
(河南康達制藥有限公司,周口 466200)
頭孢拉定(Cefradine)如圖1,化學名為(6R,7R)-7[(R)-2-氨基-2-(1,4-環己烯基)乙酰氨基]-3-甲基-8-氧代-5-硫雜-1-氮雜雙環[4.2.0]辛-2-烯-2-羧酸。性狀為白色或類白色結晶性粉末,微臭。在水中略溶,在乙醇、氯仿、乙醚中幾乎不溶。頭孢拉定為美國施貴寶公司于1972年研究成功的半合成頭孢菌素類抗生素,屬于第一代頭孢菌素類,并于20世紀70年代進入醫藥市場。它對胃酸穩定,藥代動力學良好,可同時供口服和注射使用[1-2]。其特點為耐β內酰胺酶,對耐藥性金葡菌肺炎克雷伯菌有較強的殺菌作用,對溶血性鏈球菌、肺炎球菌、大腸埃希菌、部分變形桿菌等均有抗菌作用,具有抗菌譜廣、殺菌力強、過敏反應小等優點[3-5]。臨床上主要用于治療呼吸道、泌尿道、皮膚、軟組織等感染,對胃腸道感染以及骨和關節的感染、敗血癥、心內膜炎等有較好的療效和高度安全性,因此臨床應用極為廣泛。近年來國家對輸液用抗生素管理越來越嚴格,口服類藥物越來越受到重視,頭孢拉定作為常用的口服類消炎藥,日益顯得重要。
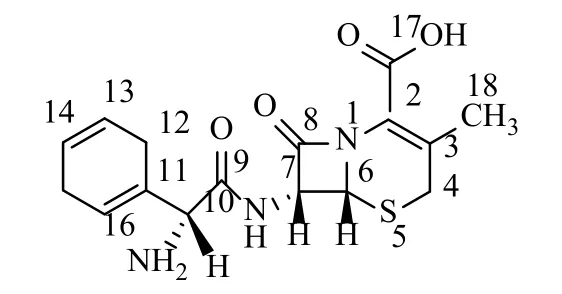
圖1 頭孢拉定
頭孢拉定的合成方法有酶法和化學法[6-13]。雖然酶法合成工藝簡單,只需一步酰化即可完成,但是收率較低,限制了此法在工業生產中的應用。因此在工業生產中一般用化學法。資料顯示的頭孢拉定工業生產多以7-氨基-3-去乙酰氧基頭孢烷酸(7-ADCA)2為起始原料,經成鹽或成酯保護后,與左旋雙氫苯甘氨酸甲基鄧鈉鹽經保護后的中間體縮合,水解得到頭孢拉定。總收率80%~90%,并且溶媒不能徹底回收,因此成本較高,不適用于工業化大生產。
在對比研究資料以后,如圖2我們采用了以2為起始原料,2與四甲基胍(TMG)成鹽,得到3(即實驗過程3.1)。左旋雙氫苯甘氨酸甲基鄧鈉鹽與特務酰氯反應成混酐4(即實驗過程3.2)后,再與3縮合得5(即實驗過程3.3),反應結束后水解得1(即實驗過程3.4)、萃取分層,然后在水層中調節pH值進行結晶、離心、烘干得到頭孢拉定晶體,二氯甲烷層用水萃取,水層與母液合并,回收制備復鹽6(即實驗過程3.5),經處理后加入下批一起結晶,收率>94%。
1 實驗儀器
日本島津SPD-10AT高效液相色譜儀;鄭州英峪予華DLSB-5/80℃低溫冷卻循環泵;江蘇金壇JJ-1定時電動攪拌;梅特勒pH計。流動相:水-甲醇-3.86%醋酸鈉溶液-4%醋酸溶液(1564:400:30:6);檢測波長254nm[4]。
2 反應路線
具體反應路線見圖2。
3 實驗步驟
3.1 7-ADCA的溶解(3)
向三頸瓶中加入二氯甲烷(水分≤0.05%,90mL,1.40mol)、2(45g,0.21mol),10~15℃加入四甲基胍(24.76g,0.21mol),攪拌反應,料液反應澄清后,15min降溫至-25℃。
3.2 混合酸酐(4)
向三頸瓶中加入二氯甲烷(水分≤0.05%,130mL,2.03mol)、4-甲基吡啶(13.4mg,0.00014mol)、DMF(70g,0.96mol)、左旋雙氫苯甘氨酸甲基鄧鈉鹽(66.0g,0.24mol),將反應瓶放置在已預冷至-35℃的低溫冷凝循環泵中降溫。攪拌下10min內加入特戊酰氯(30g,0.25mol),控制反應液溫度≤-20℃,加料完畢后攪拌反應2h,加入甲醇(0.23g,0.0072mol),三乙胺(0.73g,0.0072mol),攪拌15min,降溫至-45℃備用。
3.3 縮合反應(5)
將2的溶解液轉入到混合酸酐反應瓶中,溫度控制在-35℃~-30℃,加完后控溫-30℃反應。2.5h取樣檢測,以2的殘留量≤1.0%為反應終點結束反應,反應結束后加入甲醇(0.81g,0.025mol),三乙胺(2.56g,0.025mol),攪拌15min。
3.4 水解反應(1)
向水解反應瓶中加入純化水(195.0mL,10.82mol)、鹽酸(30%,36mL,0.34mol))。將縮合反應液轉到水解反應瓶中。用30%鹽酸調節pH值1.35~1.45,控制溫度10℃水解反應30min,觀察二氯甲烷層是否澄清。水解后轉入分液漏斗中,靜置分液。水層滴加三乙胺調pH值2.2后,真空度≤-0.09Mpa抽二氯甲烷30min。滴加三乙胺調pH值2.6,升溫至30℃繼續滴加三乙胺至pH值2.9,混濁后養晶30min,調pH值5.0,降溫0-5℃養晶2h,抽濾,抽干后濾餅用丙酮:水(30mL,0.41mol:30mL,1.66mol)洗滌,抽干后物料轉移至真空干燥箱內,控溫45±5℃,真空度≤-0.09Mpa干燥4h。得到1(59.3g,0.17mol),含量99.5%,水分4.1%,色級≤2號,質量指標高于頭孢拉定藥典基本要求。
3.5 復鹽制備
水解后二氯甲烷層加入水(60mL,3.33mol)萃取分成,水層與抽濾母液以及洗液合并,加入β-萘酚(7.08g,0.49mol),攪拌30min,用三乙胺調pH值到6.0,養晶1h,抽濾,用丙酮(50mL,0.68mol)洗滌,抽干得到復鹽濕品。在水解反應過程中加入復鹽。
3.6 溶媒回收
水解萃取分層后,二氯甲烷層精餾;洗料丙酮蒸餾;結晶母液經片堿處理后,可以回收三乙胺。經檢驗回收溶劑符合內控標準可以循環套用,不影響最終的產品質量。
4 結果與討論
在頭孢拉定的合成工藝中,最低反應溫度在-45℃,高于通常的-60℃~-75℃,降低了大量的能耗。在縮合反應結束后加入甲醇與過量的混合酸酐反應生成易于除去的酯,提高了產品質量。本方法不需要加活性炭脫色,在生產中可節省1h的脫碳時間,水層在結晶前用真空抽除二氯甲烷,使晶體能夠慢慢析出,避免暴晶,晶型過細,料黏的問題,比容大有利于制劑的分裝。通過控制攪拌速度,避免了結晶過程中凝膠等問題。
在水相中結晶、離心等后續操作,在沒有溶媒的情況下,操作更加安全,對職工危害也是最小的,并有利污水處理和環境保護。為實現工業化生產創造了更加有利的條件和環境,符合綠色生產的要求。
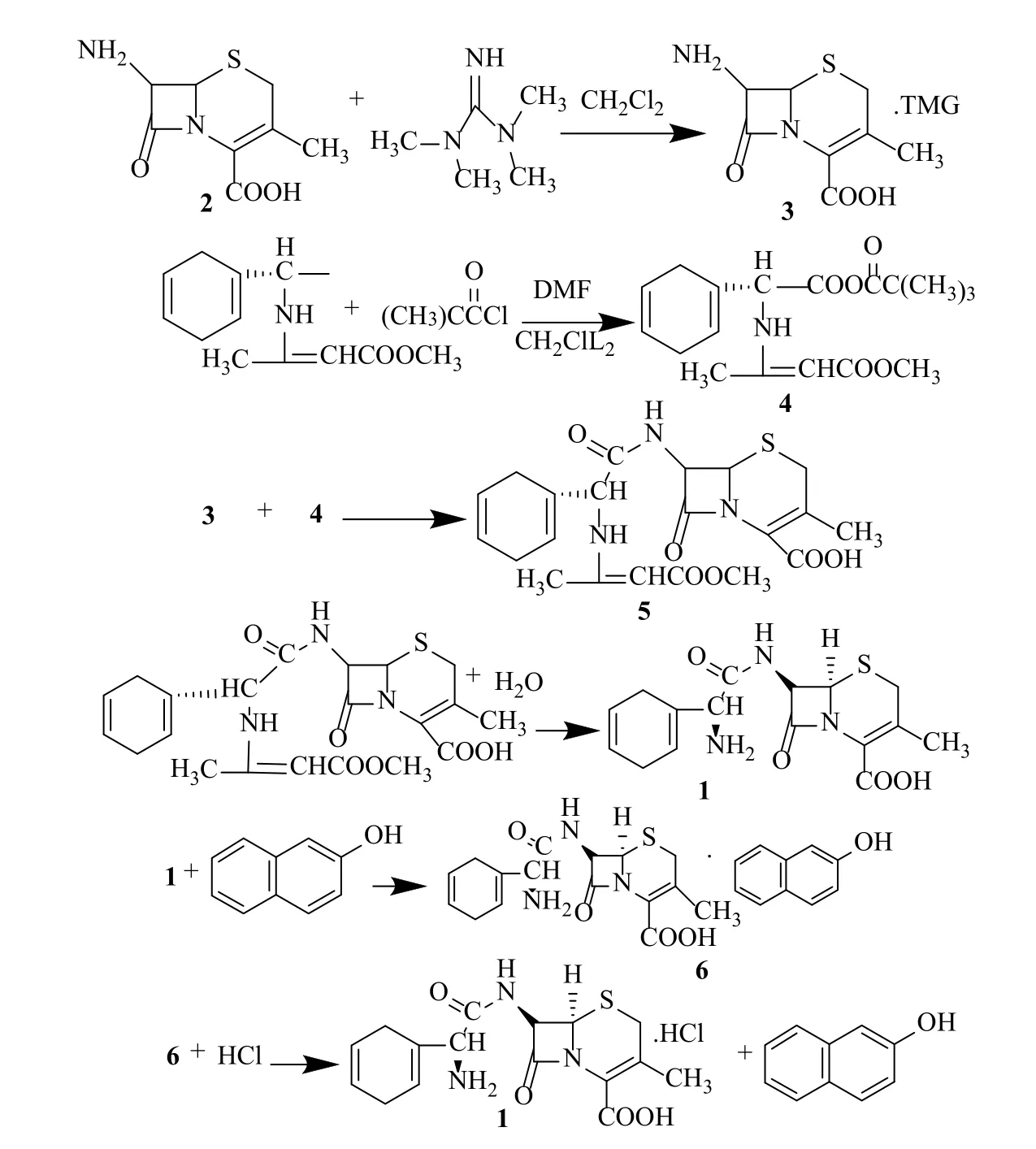
圖2 頭孢拉定合成路線圖
本方法摩爾收率由85%~90%提高到94%以上,得到的頭孢拉定色級由≥3#色降低到≤2#,含量由96%左右提高到≥99.3%,旋光+84.9°,質量指標明顯高于高于中國藥典(2015版)的要求。
1H NMR。δ:6.05(m, 1H, CH); 5.69-5.64(m, 2H, CH); 5.56-5.55(d,j=4.4Hz, 1H, CH);5.00-4.99(d,j=4.4Hz, 1H, CH); 4.54(s, 1H, CH);3.50-3.46(d,j=18.0Hz, 1H, CH); 3.12-3.071(d,j=18.0 Hz, 1H, CH); 2.73-2.66(m, 2H, CH2); 2.66-2.47(m, 2H,CH2); 1.80(s, 3H, CH3)。
13C NMR。δ169.9、168.9、163.6、131.3、126.4、126.2、123.7、122.7、121.9、58.6、58.4、56.8;28.3、26.2、24.2、18.4。
高分辨質譜分析表明樣品的準分子離子峰【M+H】+為350.1,可推斷其分子量為349.1,與1的理論分子量349.1一致,元素匹配結果表明其元素組成為C16H19N3O4S,與頭孢拉定的結構一致,證明其元素組成正確。
