玉米CMS-C同質異核、同核異質系葉綠體基因組比較分析
2019-03-11 05:23:24劉孝偉易洪楊曹墨菊
四川農業大學學報 2019年1期
邱 濤,劉孝偉,唐 津,張 鵬,易洪楊,曹墨菊
(四川農業大學玉米研究所,農業部西南玉米生物學及遺傳育種重點實驗室,成都611130)
細胞質雄性不育現象在植物界普遍存在,植物細胞質雄性不育表現為典型的細胞質遺傳。因此關于植物細胞質雄性不育分子水平的研究,主要圍繞葉綠體基因組及線粒體基因組展開。劉一農等[1]根據葉綠體DNA(cpDNA)的熱溶解曲線及含變性劑的雙向電泳分析,發現玉米CMS-C型不育系與其保持系之間的cpDNA有明顯的差異;根據雙向電泳結果發現不育系與保持系的cpDNA存在若干內切酶堿基組成的差異;根據電子顯微鏡的觀察發現不育系與保持系的葉綠體亞顯微結構方面也有明顯的區別。劉一農等[2]利用cpDNA的熱變性分析和限制性內切酶EcoRI、BamHI消化分析,根據熱變性溶解曲線及酶切片段的單向電泳和含變性劑濃度梯度的雙向電泳結果,比較分析了玉米、小麥和油菜不育系與保持系的cpDNA,并認為這3種植物的不育系與保持系的葉綠體DNA存在差異。劉祚昌[3]通過對葉綠體基因組翻譯產物分析,發現高粱不育系與其保持系之間存在差異,而甜菜不育系與保持系之間未檢測到差異。段乃彬等[4]對4個蘿卜細胞質雄性不育系和1個保持系的葉綠體基因組及線粒體基因組進行了組裝及比較分析。綜合前人的研究結果,可以發現大多數植物細胞質雄性不育可能與線粒體基因組的變異有關,某些細胞質雄性不育則可能與葉綠體基因組的變異有關。不同作物細胞質雄性不育系可能受控于不同的細胞質遺傳系統。
在DNA水平對葉綠體基因組進行研究的主要方法可歸納為3種[5]:①限制性內切酶酶切圖譜分析法。1998年以前此方法廣泛應用,通過酶切片段大小的比較,了解不同物種葉綠體基因組序列之間的變化。1980年,F.Vedel等[6]以4種麥類作物為材料,結合4種酶切圖譜分析,研究葉綠體和線粒體之間的進化關系,發現葉綠體基因組酶切多態性遠小于線粒體基因組。1983年,劉一農等[1-2]利用該法對玉米、小麥、油菜等不育系及其保持系的cpDNA進行了比較分析,并認為不育系與保持系的cpDNA存在有差異。②分子雜交和PCR法。③DNA測序技術。1986年煙草[7]和地錢[8]葉綠體基因組序列的發表使人們第一次有機會從堿基水平上對不同基因組之間進行比較,在研究方法上有了實質性的突破。之后,許多植物的葉綠體基因組測序相繼完成[9-11]。對于酶切圖譜分析法,由于酶切位點的限制,所得信息量有限,分析的局限性較大,因而將逐步被淘汰。分子雜交法主要是針對特異片段或特異位點進行分析,因此其應用也受到限制。隨著核酸測序技術的飛速發展,從全基因組水平檢測遺傳差異或多態性已經被廣泛采用。高通量測序技術(next generation sequencing)的誕生,極大地提高了核酸測序的數據量,并使得測序成本大大降低。1995年R.Maier等[12]通過構建葉綠體DNA的質粒文庫,利用雙脫氧鏈終止測序法獲得了完整的玉米葉綠體基因組序列。2015年M.Bosacchi等[13]基于細胞總DNA的二代測序數據,完成了6個玉米材料的葉綠體基因組的組裝。目前,基于混合樣品高通量測序對植物細胞器基因組進行組裝和分析的相關研究已有報道,且呈現出逐漸增多的趨勢[14-15]。
對植物細胞質基因組進行研究,傳統方法普遍需要先分離出相應的細胞器,提取其DNA,再進行測序分析。有時則需要分離出精制的細胞器,再提取其DNA進行分析。而精制葉綠體的制備,通常需要特殊的設備和復雜的操作程序。目前,基于高通量測序數據,借助于高效完善的計算機分析軟件,加之越來越多的各類核基因組及細胞器基因組測序的完成[12,16],為利用混合樣品的測序數據對細胞質基因組進行分析提供了可能和保障。利用計算機將這些大量的測序數據進行質量過濾和拼接,再通過比對參考基因組或者PCR試驗來進一步填補序列gap,最終獲得完整的細胞質基因組。
玉米細胞質雄性不育作為雜種優勢利用的重要工具以及核質互作研究的理想材料,長期以來受到遺傳學家和育種學家的廣泛關注。玉米CMS-C在生產上具有較大的應用前景。然而目前,關于玉米CMS-C及其保持系葉綠體全基因組的比較研究,尚未見報道。本研究試圖基于玉米線粒體DNA的高通量測序數據,進行葉綠體基因組組裝及分析,旨在探討該方法的可行性,并進一步了解玉米同核異質、同質異核材料之間葉綠體基因組的差異表現。
1 材料和方法
1.1 材料培養
取玉米細胞質雄性不育系C48-2、C黃早四、C698-3、C478及其保持系48-2、黃早四種子,在滅菌的培養皿上放兩層吸水紙,待無菌水浸濕,在上面均勻擺放約30粒玉米種子,再覆以一層吸水紙,用無菌水完全浸濕上層吸水紙。將培養皿置于28℃恒溫培養箱催芽2 d,再將已發芽的種子轉移到盛有營養土的盆中,并定期補充水分,培養7 d左右。
1.2 玉米線粒體分離及線粒體DNA提取
線粒體的分離和線粒體DNA的提取采用Wang J.等[17]報道的方法。
1.3 高通量測序及葉綠體基因組組裝
線粒體基因組測序由北京百邁客生物科技有限公司協助完成。對供試材料的線粒體基因組DNA分別構建文庫,然后用Hiseq2500對文庫進行雙末端測序,測序讀長(reads)的長度為125 bp。為了使得后續分析的結果更加準確可靠,對原始的測序數據進行如下處理:①過濾某個位點N含量≥80%的所有reads;②截取read 1、read 2中高質量區域序列;③過濾低質量的reads;④過濾N含量大于10%的reads;⑤過濾當adapter序列與reads比對上15 bp或以上且錯配數≤3的reads;⑥當一對reads完全比對上其他的reads,過濾此冗余的reads。
本試驗分離提取的玉米線粒體DNA為粗制線粒體DNA,含有一定量的葉綠體DNA,故本研究通過以已發表在NCBI上的玉米葉綠體基因組為參照序列,利用Bowtie2(http://bowtie-bio.sourceforge.net/index.shtml),參數按軟件默認設置,得到所需的sam文件。然后用perl語言編寫腳本,將提取序列中與參考序列匹配的reads,生成fastq格式文件,用于后續的拼接。短序列比對獲取測序結果中葉綠體DNA的reads。利用Velvet軟件對完成過濾和比對后的reads進行組裝,通過尋找短序列之間的重疊區域(overlap)將高質量的短序列拼接成重疊群序列(contig),然后將所有的短序列定位到拼好的重疊群序列(scaffold)上,再根據PE(pair-end)關系將重疊群連接成scaffold序列。因參數設置對Velvet的運行結果有很大的影響,尤其是K-mer值和覆蓋深度(coverage)的設置。所以實驗設置了多個參數進行調試,再將組裝序列用BlastN比對到參考基因組,以此對組裝獲得的scaffold序列進行排序。在scaffold序列內,根據gap上下游序列設計引物并進行PCR擴增,利用Sanger法測序擴增產物,并將測序結果與組裝序列和參考序列進行比較,以此填補組裝scaffold序列內的gap序列,不同scaffold間的序列以同樣的方法進行拼接,最終得到完整的環狀玉米葉綠體基因組。
1.4 玉米葉綠體基因預測及注釋
采用在線注釋軟件DOGMA(http://dogma.ccbb.utexas.edu/)對2份玉米CMS-C不育系和2份保持系的葉綠體基因組完整序列進行基因預測和基因功能分析,根據起始密碼子和終止密碼子序列手工調整DOGMA初步注釋的編碼蛋白基因范圍,并根據CpBase數據庫進行了基因注釋,通過BLAST比對參考基因組和手工校準相應基因信息。
功能注釋使用同源比對的方法,通過基因預測得到樣品的氨基酸序列,與已知的蛋白數據庫進行比對,把供試材料的基因和其相對應的功能注釋信息結合起來,得到注釋結果,以推測它們的結構、功能以及進化上的聯系。由于每一條序列比對結果可能超過一條,為保證其生物意義,注釋時保留一條最優比對結果作為該基因的注釋。
2 結果與分析
利用Nano Drop 2000檢測玉米線粒體DNA質量,選取質量合格的樣品,送北京百邁客生物科技有限公司,使用Hiseq 2500測序平臺對樣品進行測序分析。對48-2、C48-2、黃早四和C黃早四4個材料的線粒體DNA分別構建文庫,進行雙末端測序,Reads長度為125 bp。對Illumina平臺Hiseq 2500測序系統產出的原始數據進行整理(表1),原始文庫插入片段為500 bp,獲得的測序數據量均超過1.2 Gb,測序深度均超過2 000×。將低質量的reads以及完全相同的冗余reads過濾后,Clean Data數據量均大于1 Gb,Q20值大于90%,Q30值大于85%。

表1 供試材料粗制線粒體DNA的測序結果Table 1 mtDNA sequencing result informations of 4 experimental materials
2.1 玉米CMS-C不育系及保持系葉綠體基因組的組成
通過拼接組裝分別獲得2個不育系及2個保持系的葉綠體基因組序列,C48-2葉綠體基因組大小為140 473 bp,C黃早四為140 478 bp,48-2為140 458 bp,黃早四為140 448 bp,GC含量均為38.4%(表2)。基因組大小與已報道的玉米葉綠體基因組大小相近。采用在線注釋軟件DOGMA(http://dogma.ccbb.utexas.edu/)對2份玉米CMS-C不育系和2份保持系的葉綠體基因組完整序列進行基因預測和基因功能分析,并根據CpBase數據庫進行基因注釋,其中蛋白編碼基因均為84種,tRNA基因均有30種,rRNA基因均有4種。組裝的葉綠體基因組均由2個反向重復序列和1個長單拷貝序列及1個短單拷貝序列組成。重復序列含有4種rRNA基因及部分tRNA基因和蛋白編碼基因,故整個葉綠體基因組的rRNA基因有8個,tRNA基因有48個或49個,4份材料的蛋白編碼基因分布在117至122之間,數目不等。部分基因在不同胞質材料中存在多拷貝現象。
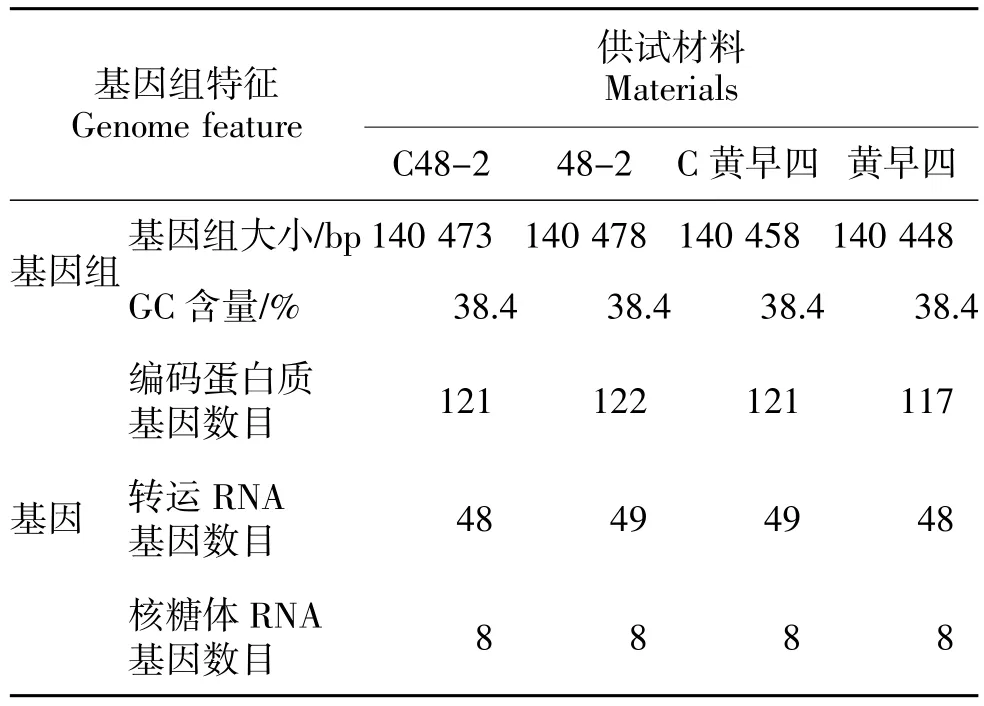
表2 4份玉米葉綠體基因組的大小及基因組成Table 2 The size and gene content of novel assembly 4 maize chloroplast genomes
C48-2、C黃早四、48-2和黃早四4個葉綠體基因組均注釋到84種編碼基因。不育系C48-2、C黃早四以及保持系48-2葉綠體基因組中的atpF、ndhA、orf42、rps15、rps19、rps7、ycf15、ycf68基因均有2個拷貝,rpl23、rps12、ycf3基因有3個拷貝,ndhB和rpl2有4個拷貝,orf56和ycf2存在6個拷貝,拷貝數最多的基因是ycf1,共存在8個拷貝,其余基因均為單拷貝;黃早四中rps19只有1個拷貝,rpl2和rpl23只有2個拷貝,其余基因拷貝數均與其他材料相同。4個葉綠體基因組中除了注釋到84種蛋白編碼基因外,還預測到6個假基因ycf1,ycf2,ycf3,ycf4,ycf15,ycf68,分別以單拷貝或多拷貝存在,拷貝數最高可達8個。
組裝的4個玉米葉綠體基因組中均含有30種tRNA基因,且不同的tRNA基因其拷貝數也不同。C48-2、C黃早四、48-2和黃早四4個葉綠體基因組中的trnfM-CAU、trnL-CAA、trnL-UAA、trnM-CAU、trnN-GUU、trnV-GAC、trnV-UAC基因均存在2個拷貝;trnT-GGU存在3個拷貝;trnA-UGC存在4個拷貝;trnl-CAU存在6個拷貝。trnK-UUU在C48-2中僅有1個拷貝,而在其他材料中存在2個拷貝;trnH-GUG在黃早四中存在1個拷貝,在其他3個材料中有2個拷貝。4個供試材料的葉綠體基因組中均有4種rRNA基因,分別為rrn4.5、rrn5、rrn16和rrn23,且都為2個拷貝。
2.3 不育系與保持系葉綠體基因組比較
對C48-2、C黃早四、48-2和黃早四的葉綠體基因組進行序列同源性比較和系統進化分析發現,兩個不育材料C48-2、C黃早四的葉綠體基因組間序列基本相同,48-2與不育材料進化距離較近,而黃早四與不育材料進化距離相對較遠。基于4個葉綠體基因組共線性分析,可以發現不同材料間葉綠體基因組高度保守(圖1、圖2)。
圖1的結果表明,來源相同的2個不育細胞質材料具有較大的一致性,而2個保持系之間的差異大于同質異核不育系之間的差異,說明核背景對細胞質基因組的影響較小。圖2的結果顯示,2個不育材料葉綠體基因組間序列基本相同,不同材料間葉綠體基因組高度保守。
利用Codon Code Aligner軟件比對分析同核異質玉米葉綠體基因組間的變異信息,篩選2組不育系及保持系中共有的SNP和InDel位點。C48-2與48-2比對檢索到29個SNP和21個InDel位點,C黃早四與黃早四比對共檢索到42個SNP位點和24個InDel位點。比較這兩組同核異質材料變異信息,統計共有的SNP和InDel位點見表3,從表3可以看出,大多數的堿基變異和全部的插入、缺失都發生在基因組的重復序列。

圖1 4個玉米材料葉綠體基因組系統進化分析Figure 1 Phylogenetics analysis of chloroplast genomes among four maize materials
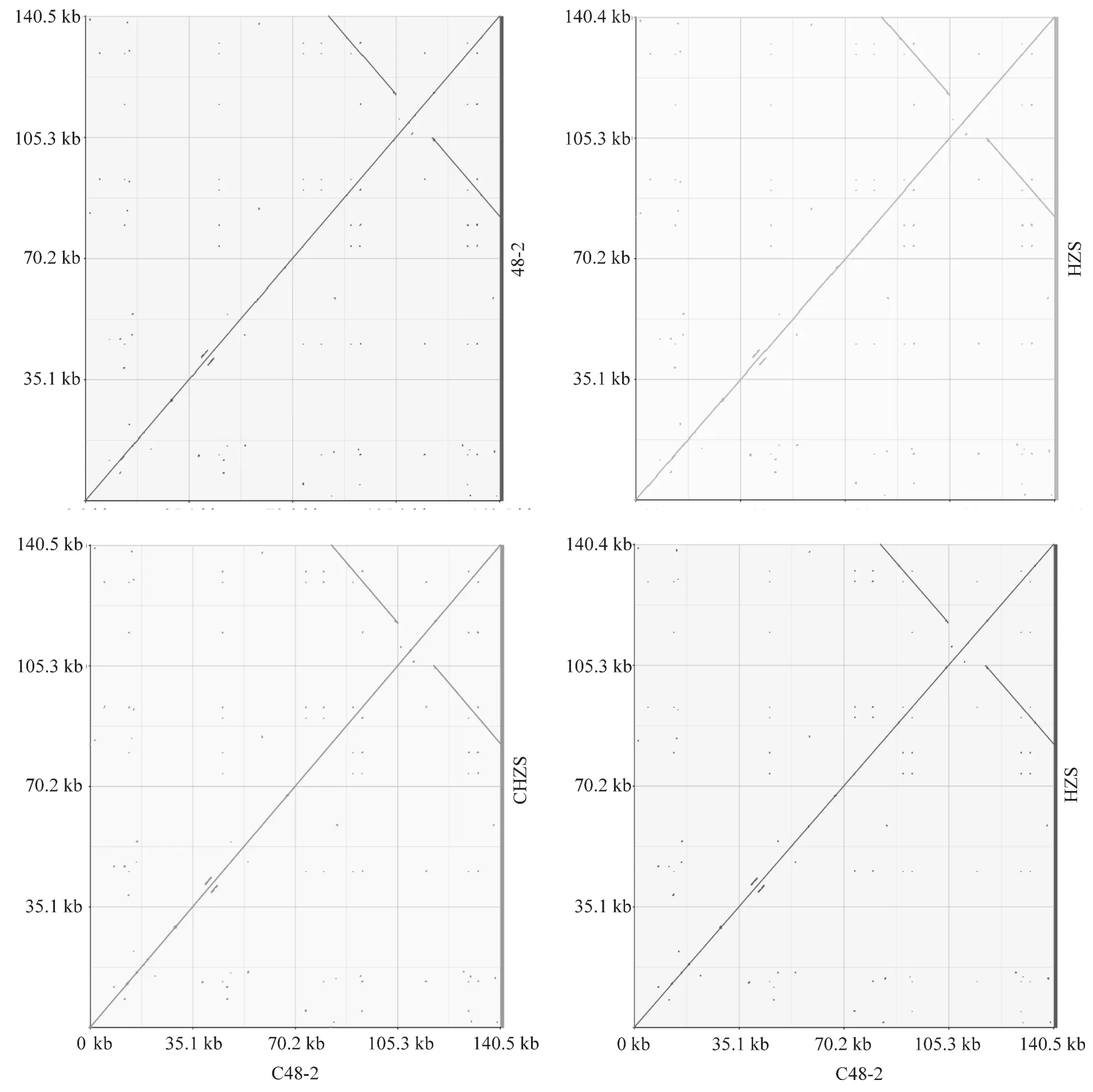
圖2 4個玉米材料葉綠體基因組共線性分析Figure 2 Synteny analysis of chloroplast genome among four maize materials

表3 兩套同核異質系葉綠體基因組SNP和InDel共有位點統計Table 3 The common SNP and InDel sites of chloroplast genome between CMS-C lines and maintain lines
本試驗利用WebSNAPER(https://pga.mgh.harvard.edu/cgi-bin/snap3/websnaper3.cgi)對檢索到的變異位點設計引物,然后以CMS-C不育系及其保持系DNA為材料進行PCR擴增驗證。基于上述策略最終設計出S8-C和S8-N兩對額外錯配堿基引物(表4),兩對引物均能有效區分CMS-C不育細胞質和正常細胞質(圖4)。
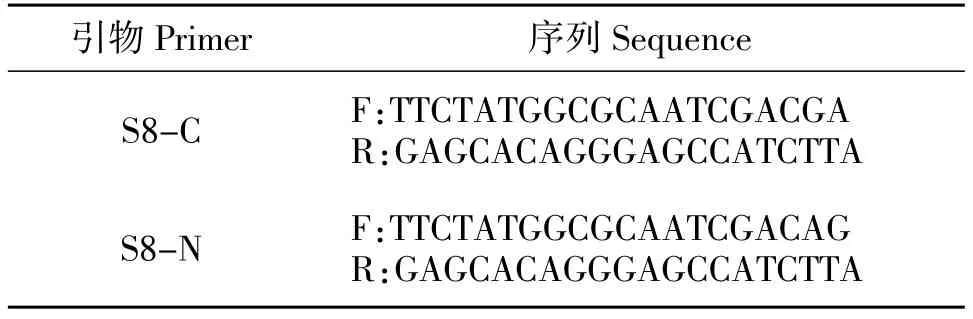
表4 基于SNP設計的兩對引物序列Table 4 The primer sequences of S8 based on SNP

圖4 利用標記S8-C和S8-N對不同細胞質類型進行PCR擴增Figure 4 The PCR amplification results of chloroplastid SNP markers S8-C and S8-N for different cytoplasmic materials
3 討論與結論
3.1 基于粗制線粒體的高通量測序數據可以完成葉綠體基因組的組裝
植物細胞含有葉綠體、線粒體和細胞核3套不同的遺傳系統。一般情況下所提取的細胞總DNA,實際上是一個既包含核基因組DNA也包含細胞質基因組DNA的混合體。目前基于總DNA混合樣品的高通量測序數據,已經在某些作物上完成了線粒體基因組或葉綠體基因組的組裝。本研究通過利用差速離心制備粗制線粒體,目的在于有效富集線粒體、葉綠體等細胞器遺傳物質,盡可能排除細胞核遺傳物質。有資料報道[18],每個玉米葉片細胞中葉綠體DNA大約有3 000~4 000個拷貝,而相應線粒體DNA的拷貝數量僅為30~100。這就為利用粗制線粒體DNA的高通量測序結果,分析葉綠體體基因組提供了理論基礎。本研究成功組裝出C48-2、C黃早四、48-2以及黃早四4個葉綠體基因組,通過與已經測序的C-B37和N-B37葉綠體DNA序列進行比較[13],發現在基因組大小、結構以及基因組成等方面有較大的一致性,說明基于線粒體基因組的高通量測序數據可以有效組裝出葉綠體基因組。本研究組裝的4個葉綠體基因組大小與1995年首次完成的玉米葉綠體全基因組較為一致[12];并且均含有84種蛋白質編碼基因、30種tRNA基因、4種rRNA基因,與1995年的報道的tRNA基因、rRNA基因數量完全一致,僅在蛋白質編碼基因的數量上存在差異。
早期對葉綠體基因組的研究,通常需要分離出葉綠體,而葉綠體的分離不僅復雜繁瑣且受實驗設備限制,一定程度上制約了葉綠體基因組的研究進展。隨著計算機分析軟件發展、算法的改進及測序方法的完善和提升,利用混合樣品測序數據,可同時開展不同遺傳體系的基因組組裝分析,比如利用細胞總DNA的高通量測序數據,可同時開展細胞核基因組、葉綠體基因組和線粒體基因組的組裝分析,這樣既省去了葉綠體和線粒體細胞器的分離純化過程,也提高了實驗效率,使得對線粒體、葉綠體基因組的研究不再受制于細胞器的分離[19]。
3.2 玉米CMS-C不育系與保持系的葉綠體基因組具有較高的一致性,但也存在一定的差異
通過對同質異核和同核異質葉綠體基因組進行序列同源性比較和系統進化分析,發現具有正常細胞質的黃早四與48-2葉綠體基因組之間的序列相似性要小于同質異核不育系C48-2與C黃早四之間的相似性。這與2015年M.Bosacchi等[13]的報道結果極為一致,即玉米CMS-C、CMS-S、CMS-T這3類不育胞質之間葉綠體基因組的相似性小于來自于瑞德黃馬牙群(reid yellow dent)的自交系B73(選自BSSSC2)與B37(選自BSSSC0)之間葉綠體基因組的相似性。通過對本研究組裝的玉米葉綠體基因組進行分析,盡管不同材料間玉米葉綠體基因組具有高度的保守性,然而它們之間卻仍然存在一些SNP位點。這與早期人們通過酶切圖譜分析或熱變性溶解曲線分析,所得的結果較為一致,即不育系與保持系的葉綠體DNA在堿基組成上有差異[1-2]。這些結果一致說明不育系與保持系的葉綠體DNA存在堿基組成上的差異。
3.3 成功開發出可以區分玉米CMS-C不育胞質與正常胞質的葉綠體特異SNP標記
比較本研究獲得的兩套同核異質材料葉綠體基因組間的共有的SNP差異,并和已經測序的玉米N-B37、C-B37葉綠體基因組相比,篩選到不育細胞質CMS-C與正常細胞質之間共有的SNP位點。基于這些共有的SNP位點,成功設計出能夠區分玉米CMS-C不育細胞質和正常細胞質的葉綠體特異引物。
