聚苯乙烯多孔吸附微球的制備
2019-02-26 07:56:08杜中杰張秀生
中國塑料 2019年2期
關鍵詞:結構
顧 玥,段 成,杜中杰,,張秀生,張 晨,*
(1. 北京化工大學材料科學與工程學院,碳纖維及功能高分子教育部重點實驗室,北京 100029;2. 北京化工大學常州先進材料研究院,江蘇 常州 213164;3. 北京中科太康科技有限公司,北京 100176)
0 前言
血液灌流技術由于療效顯著被作為新的血液凈化方式并廣泛應用,該技術的核心吸附材料需要滿足吸附性能優異、對人體無毒無害、化學性質穩定、機械強度良好、血液相容性好等要求[1]。離子交換樹脂最早被用作吸附材料,但其選擇性低、吸附效率差;之后出現的活性炭吸附材料,存在血液相容性差、炭粒易脫落引起栓塞等問題[2-4]。聚苯乙烯基多孔吸附樹脂有良好的生物相容性、化學穩定性和機械強度,被看作是新一代血液灌流凈化材料的首選。但是聚苯乙烯結構的疏水性以及常規聚苯乙烯大孔微球中孔徑尺寸過大則成為了開發聚苯乙烯基血液凈化材料需要解決的關鍵問題[5-6]。
本實驗首先選用聚苯乙烯作為樹脂基體,二乙烯基苯為交聯劑,采用懸浮聚合方法得到聚苯乙烯微球,并且在配方體系中加入致孔劑甲苯以及共聚單體馬來酸酐,即可得到含有大孔隙的親水改性聚苯乙烯微球(MPSS)[6]。參考文獻報道[7-10],利用付-克超交聯反應在聚苯乙烯微球內形成介孔,有效地提高了微球的比表面積。所制備的高比表面積聚苯乙烯微球(HCLPS)以戊巴比妥鈉為樣本進行了吸附性能測試,研究了介孔結構與馬來酸酐用量對HCLPS的吸水性及戊巴比妥鈉吸附性能的影響。
1 實驗部分
1.1 主要原料
苯乙烯(St)、二乙烯基苯(DVB)、MAH、聚乙烯醇(1799)、分析純、無水氯化鋁(AlCl3),分析純,天津光復精心化工研究所;
2,2-偶氮二異丁腈(AIBN),分析純,九鼎化學;
甲苯、二氯甲烷(DCM)、無水甲醇、無水乙醇、丙酮,分析純,北京化工廠;
氫氧化鈉,分析純,江蘇海安石化廠;
苯乙烯和二乙烯基苯單體經過減壓蒸餾除去阻聚劑后使用,2,2-偶氮二異丁腈經過重結晶后使用。
1.2 主要設備與儀器
電子天平,FA2004 ,上海良平儀器儀表公司;
恒速電動攪拌機,JB90-SH,上海標本模型廠;
電熱鼓風干燥箱,DHO9053,吳江韻達烘箱設備有限公司;
恒溫水浴振蕩器,KQ5200E,昆山市儀器有限公司
傅里葉紅外變換光譜儀(FTIR),Nicolet-Nexus 670,美國Thermo公司;
掃描電子顯微鏡(SEM),S4700,日本日立公司;
X射線光電子能譜分析(XPS),ESCALAB 250,美國Thermo Fisher Scientific公司;
氣體吸附儀,JW-BK122W,北京精微高博科學技術有限公司。
1.3 樣品制備
MPPS微球的制備:12.00 g苯乙烯、2 g二乙烯基苯、0.80 g偶氮二異丁腈與1.20 g甲苯混合后加入裝有機械攪拌、冷凝管、溫度計的250 mL三口燒瓶中,升高溫度至45 ℃后加入一定量的馬來酸酐,在一定轉速下加入20 mL的1.5 % PVA水溶液,然后加入130 mL去離子水;升溫至70 ℃,當聚合物顆粒變硬即可結束反應[11];將產物用布氏漏斗抽濾,用熱水清洗3次后放入真空烘箱中干燥得到MPSS微球;將馬來酸酐含量占苯乙烯單體比例0、10 %和20 %的樣品分別命名為F01、F02、F03;
HCLPS微球的制備:1.00 g MPPS微球、50 mL二氯甲烷和8.00 g無水氯化鋁加入250 mL單口燒瓶,室溫條件下在磁力攪拌下攪拌12 h使微球充分溶脹,然后再在70 ℃下反應12 h;將微球分離出來,用丙酮和甲醇分別洗滌除去無水氯化鋁;然后將微球在鹽酸∶去離子水∶丙酮=5∶3∶2的100 mL溶液中浸泡24 h,再經甲醇索氏抽提12 h,置于150 ℃真空烘箱中2 h后得到HCLPS[12];對應不同MPSS微球得到的樣品分別命名為HF01、HF02、HF03。
1.4 性能測試與結構表征
FTIR測試,將待測樣品磨成粉末,溴化鉀按照1∶100的比例混合均勻后壓片,在400~3 600 cm-1的范圍內測定;
SEM觀察樣品微觀形貌,樣品的斷面進行噴金處理后觀察,其中空腔和通孔尺寸采用Nano Measure軟件進行分析和統計;
XPS分析材料中表面元素組成;
采用77 K下的氮氣吸脫附曲線通過Brunauer-Emmett-Teller理論計算比表面積,測量前將樣品于150 ℃下真空脫氣100 min;
吸水性測試:在精密分析天平上稱量定性濾紙重量,將濾紙折成紙包,取一定量的微球樣品放置于紙包內,封好并稱重,記錄數據;將紙包浸泡于去離子水中12 h(確保紙包完全浸于水中),12 h后取出紙包,靜置,待紙包無流動態水時,稱量紙包;取出紙包內的微球,再次稱重;通過計算得出微球吸水前后的重量,即可算出微球的吸水性;
戊巴比妥鈉吸附性能測試:配制80 mg/L濃度的戊巴比妥鈉溶液,取25 mL置于50 mL具塞錐形瓶中,稱取1.0 g吸附劑(干重)投入瓶中,置于(37±1)℃以(60±10)次/min的速率在恒溫水浴振蕩器內振蕩2 h,用紫外分光光度法于240 nm處測量計算吸附前后的戊巴比妥鈉溶液濃度,按式(1)計算下降率[13]。
(1)
式中C——溶液下降率, mg/L
C0——吸附前溶液濃度, mg/L
Ct——吸附2 h后溶液濃度, mg/L
2 結果與討論
2.1 FTIR和XPS表征
圖1是樣品F01和F02以及超交聯后樣品HF02的FTIR譜圖和XPS全光譜圖。首先在FTIR譜圖中,可以看出純PS的F01樣品中,出現了3 075 cm-1處苯環中C—H的伸縮振動峰,而在加入10 % MAH的F02樣品中出現了1 733 cm-1處羰基的吸收振動峰,表明MAH和St單體發生了共聚,所合成的MPPS微球中含有親水的酸酐基團。將F02樣品進行超交聯后得到產物HF02,其FTIR譜圖中1 690 cm-1和1 712 cm-1處的峰為MAH中羰基的特征吸收峰,而2 935 cm-1處亞甲基中C—H伸縮振動峰明顯強于F02,表明在微球內發生了付-克烷基化反應。另外在3 475 cm-1處出現了順丁烯二酸中羥基的振動峰,說明有部分MAH發生了水解。
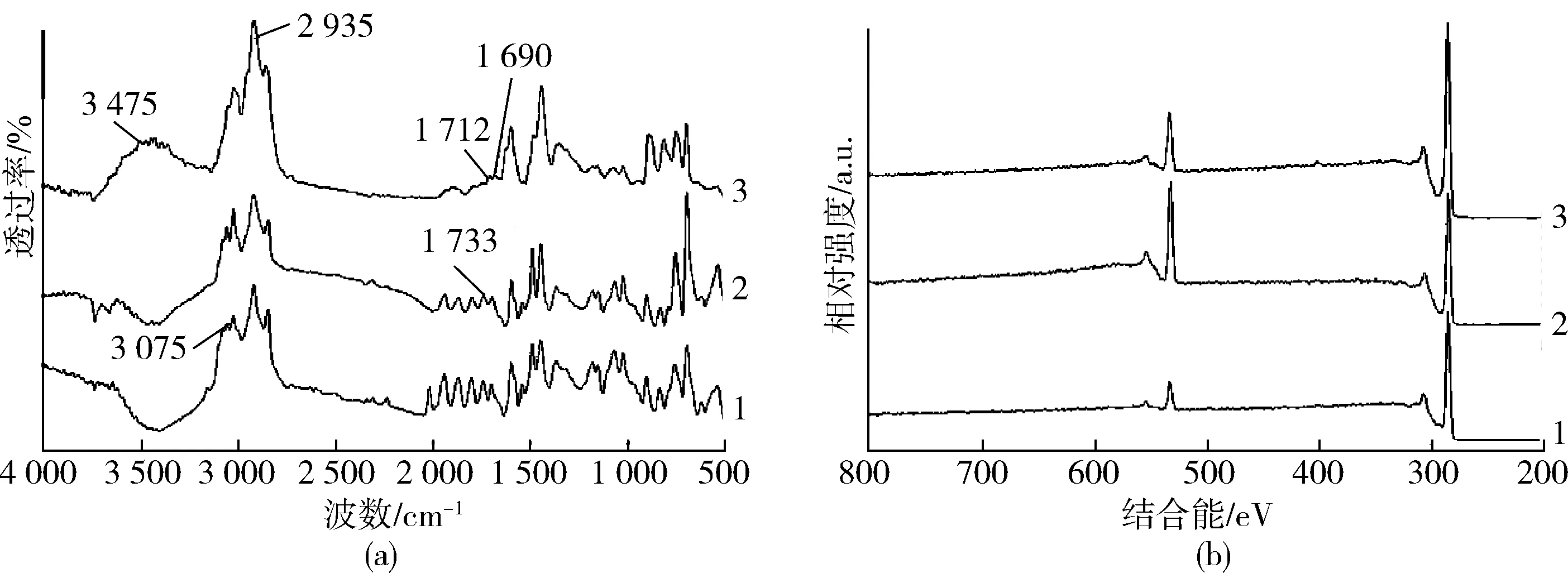
1—F01 2—F02 3—HF02(a)FTIR譜圖 (b)XPS譜圖圖1 F01,F02與HF02的FTIR和XPS譜圖Fig.1 FTIR and XPS spectra of F01, F02 and HF02
對比3個樣品的XPS全光譜圖和對應樣品中C和O的元素含量(表1),可以看出在聚合體系中加入MAH后,產物F02的O元素含量明顯升高(F01樣品中少量O元素可能來自于分散劑聚乙烯醇)。而在超交聯處理后HF02樣品的O元素含量有所減少,是由于發生超交聯反應中引入大量C元素。
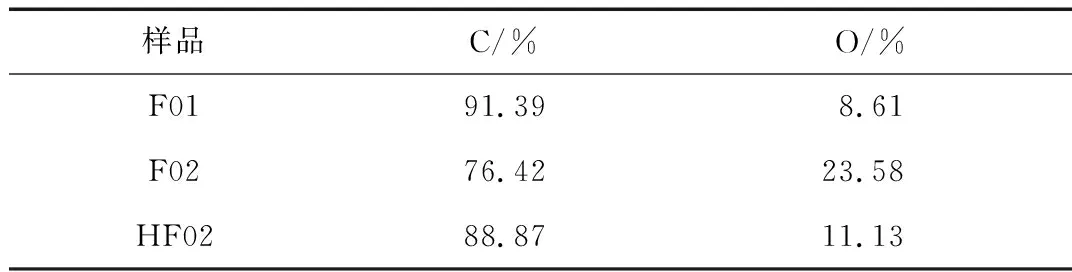
表1 F01、F02和HF02的C、O含量
綜合以上結果,可以說明在MPPS微球中引入了親水基團,并且成功地進行了超交聯反應。
2.2 SEM分析
采用掃描電子顯微鏡觀察微球的形貌(圖2),從圖2(a)中可以看到MPPS微球的形貌基本完整,球形度高,其粒徑在2 mm左右。對比可以發現,馬來酸酐的加入使F02微球的球形度降低,粒徑減小至1 mm左右。造成這種變化的原因可能是由于馬來酸酐部分水解后容易向水相中遷移,水油兩相間的物質交換增多導致液滴穩定性變差,懸浮體系中的液滴尺寸也隨之降低,但液滴也因此容易變形[14]。交聯后微球的粒徑又增大到2 mm,這說明溶脹后進行的超交聯反應,在聚苯乙烯分子鏈之間通過化學反應引入了橋聯結構,在形成微孔與介孔的同時,微球的外觀尺寸也略有膨脹。除此之外,還可以注意到HF02微球的表面出現了明顯裂痕。這是因為在超交聯反應后,聚苯乙烯分子鏈運動困難,因此在脫除溶劑的過程中產生較大的內應力,使微球表面發生破裂[2]。
2.3 BET測試
采用BET方法測試了樣品的微孔結構(圖3)。F01樣品在較低相對壓力的條件下(P/P0<0.01),未表現出氮氣吸附,說明采用懸浮聚合方法制備的聚苯乙烯微球中不存在微孔結構。而經過超交聯處理后,樣品HF01、HF02與HF03均在P/P0<0.01的條件下表現出了較高的氮氣吸附量,這是具有微孔結構的材料的特點[15],說明通過超交聯反應能夠在聚苯乙烯微球中有效地制造出微孔結構。這主要是由于二氯乙烷中的氯原子與聚苯乙烯的苯環反應后,顯著提高了聚合物網絡的交聯度。在移除溶劑后,聚合物網絡中原本由溶劑占據的部分即會成為微孔結構[16]。
通過吸脫附等溫線計算出樣品的比表面積和孔體積,結果列于表2。在進行超交聯處理之前,聚苯乙烯微球的比表面積和總孔體積分別只有33.5 m2/g以及0.093 cm3/g,其中的孔隙結構主要是由于在制備過程中加入了致孔劑甲苯而造成。經過超交聯處理后,微球的比表面積明顯增加,最大可以達到570.9 m2/g,正是由于超交聯反應在聚合物中形成微孔結構所導致。進一步對比經過超交聯處理的不同馬來酸酐含量的聚苯乙烯微球樣品,發現微球的比表面積以及微孔體積隨著馬來酸酐加入量的增加而減小。這可能是由于在分子鏈中引入的馬來酸酐單元不參與傅克反應,因此分子鏈的交聯度會隨著馬來酸酐用量的增加而降低,因此形成的微孔孔體積和比表面積也隨之降低[17]。
2.4 吸水性測試
不同結構樣品的吸水性如圖4所示,可以看出,經過超交聯后微球的吸水量明顯提高。這應該是由超交聯處理后微球中微孔能夠容納更多的水分子[18]。同時,也可以看出不論是否經過超交聯處理,微球的吸水量都會隨著馬來酸酐加入量的增加而增加,這可以歸因于馬來酸酐基團良好的親水性。因此 微球中的馬來酸酐含量越高,聚合物的親水性越好,吸水的能力就越強。總之,所制備的微球具有良好的親水性能,可以很好在水性介質中進行吸附。
2.5 戊巴比妥鈉吸附性能測試
取已知含水量的濕態HF01~HF03微球樣品1 g,并折合成干重后,按戊巴比妥鈉吸附測試方法測得吸附后濃度Ct,計算出下降率。其中初始濃度C0=80 mg/L,由此得出的下降率C就是戊巴比妥鈉吸附率,測試結果列于表3。首先,微球對戊巴比妥鈉的吸附率最高可達91.90 %,說明所制備的微球能夠有效地脫除戊巴比妥鈉。其次,超交聯處理后微球的戊巴比妥鈉吸附率(HF01)明顯升高,說明經過超交聯反應在微球中引入的微孔結構對于戊巴比妥鈉的吸附具有決定性影響。這是由于微孔結構能夠與客體分子產生較強的相互作用力并為材料提供較高的比表面積[19];第三,微球的戊巴比妥鈉的吸附能力會隨著馬來酸酐含量的增加而增加,我們認為微球中馬來酸酐基團以及由馬來酸酐水解而得的羧酸基團能夠使微球材料表面帶電,從而增強了微球吸附戊巴比妥鈉的能力[20]。最后,盡管HF03并沒有最高的比表面積,但是卻表現出了最高的吸附率,由此可以推論出只有合理控制微孔結構與特定基團,才能夠有效提高微球的戊巴比妥吸附性[21]。
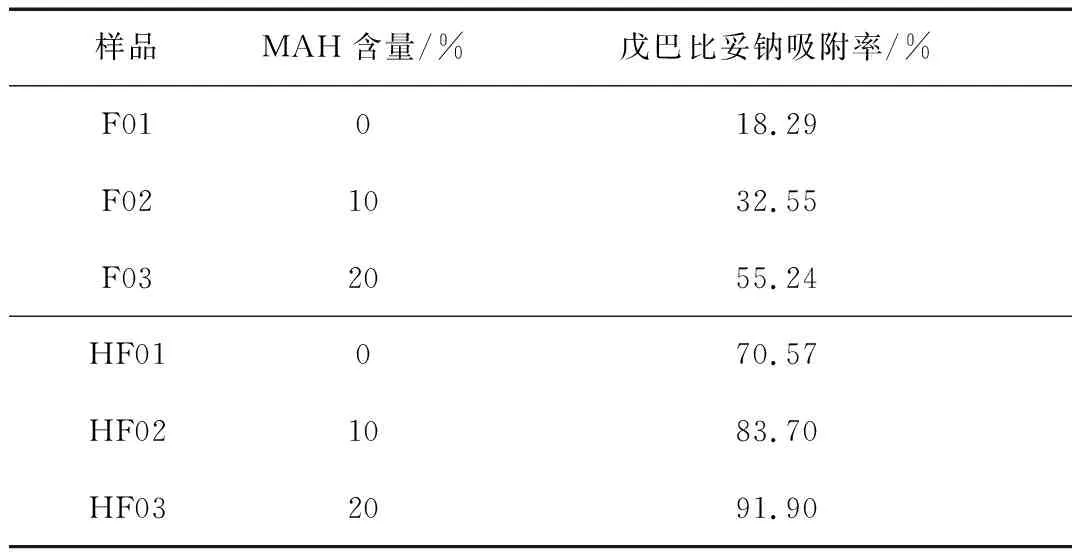
表3 HCLPS微球的戊巴比妥鈉吸附率測試結果
3 結論
(1)通過懸浮聚合方法成功制備了粒徑為1~3 mm、含有極性親水基團的交聯聚苯乙烯多孔微球,再對其以二氯甲烷為交聯劑,無水氯化鋁為催化劑進行超交聯處理,制備得到的HCLPS微球比表面積值在200~600 m2/g之間;
(2)微球的吸水性以及戊巴比妥鈉吸附性能都會隨著微孔的形成以及馬來酸酐的引入而得到改善,當馬來酸酐的含量為20 %時, HCLPS微球的戊巴比妥鈉吸附率可以達到91.9 %;繼續增加馬來酸酐的含量會嚴重影響微球的球形度與形貌,并且在超交聯處理后形成的微孔結構也有限。
猜你喜歡
小獼猴智力畫刊(2023年4期)2023-04-23 08:49:58
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
中學生數理化·高一版(2018年1期)2018-02-10 05:20:03
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
七彩語文·寫字與書法(2016年7期)2016-07-28 21:40:22
七彩語文·寫字與書法(2016年6期)2016-07-15 19:36:34
人間(2015年21期)2015-03-11 15:23:21
現代企業(2015年9期)2015-02-28 18:56:50
