HPLC法對黃精顆粒和提取物的色譜研究
2019-02-11 10:30:22孫艷平王榮楊寬申旭霽汪興軍
貴州醫藥 2019年12期
關鍵詞:標準
孫艷平 王榮 楊寬 申旭霽 汪興軍
(西安醫學院藥學院,陜西 西安710021)
黃精(P sibiricum Red.)是臨床上重要的中藥材,全球約有50多種,主要分布在北溫帶、北亞熱帶,而我國具有31種,多分布在南方熱帶以外地區[1]。《中國藥典》2010版收載的黃精原植物有三種:黃精(P.sibiricum Red.)、多花黃精(P.cyrtonema Hua)、滇黃精(P.kingianum Coll.et Hemsl)。黃精的藥用部位是根莖,能補氣養陰、健脾、潤肺、益腎作用[2]。用于脾胃虛弱,體倦乏力,口干食少,肺虛燥咳等癥狀。黃精身份相對較多,包括:木質素、多糖、氨基酸、皂苷類、微量元素等,且以多糖、皂苷類含量較高[3]。現代藥理 結 果 表 明[4-5]:黃 精 具 有 抗 衰 老、調節機體免疫、改善記憶、抗腫瘤等作用。黃精顆粒屬于常用的中成藥,且與傳統藥物相比具有藥物使用方便、安全性高等特點,但是臨床上對于其藥物成分缺乏統一的質量標準[6]。高效液相色譜法簡稱HPLC,是在經典液相色譜法的基礎上,引入了氣相色譜法的理論和實驗技術,以高壓輸送流動相,采用高效固定相及高靈敏度檢測器,發展而成的現代液相色譜分析方法[7]。本研究以黃精顆粒和提取物為對象開展研究,探討HPLC 在黃精顆粒和提取物的含量測定效果。報道如下。
1 材料與方法
1.1 (1)儀器:托盤天平;分析天平;碾槽;量筒;燒杯;玻璃棒;紗布;蒸發皿;標準篩;砂鍋;Agilent Technologies 1220Infinity LC高效液相色譜儀,購于美國安捷倫科技有限公司;Agilent HC-C18(2)色譜柱(4.6 mm×250 mm,5μm),KQ-250B 型超聲清洗器,購于昆山市超聲儀器有限公司;容量瓶;移液管;注射器;0.45μm 微孔濾膜;進樣針。(2)試劑為黃精粉末;黃精顆粒;色譜乙腈(美國制造)(批號:20130318);薯蕷皂苷元標準品(中國藥品生物制品檢定所)(批號:201403181);純凈水。
1.2 黃精顆粒及提取物的制備 (1)黃精顆粒制備:取黃精藥材100g于碾槽中碾碎取出,放入砂鍋中加入800 mL 的自來水,浸泡20 分鐘,煎煮30min,過濾除雜,濾渣加入400 mL 自來水,煎煮30min,過濾除雜,合并濾液,濃縮至濃度為1.1左右(90~100mL),換到水浴鍋繼續濃縮至玻璃棒挑起成絲,稱量,記錄浸膏的量,趁熱加入蔗糖邊加邊攪至成團,慢慢加入糊精邊加邊制成握之成團,觸之即散狀態,過16目篩,過篩后的顆粒在60-80 ℃烘箱中干燥,整粒,備用[8]。(2)黃精顆粒提取物制備:分別精密稱取黃精顆粒7g(相當于原藥材2g)和藥材粉末2g,置具塞錐形瓶中,按料液比為1∶20加入80%的乙醇,超聲提取2次,溫度60 ℃,時間50min,過濾,合并濾液,蒸干,加入20mL 蒸餾水溶解,加入20 mL 的正丁醇萃取二次,正丁醇層置于具塞錐形瓶中保存;向錐形瓶中加入少量甲醇溶解,分別置于10mL的容量瓶中,加入甲醇定容,置于2 ℃冰箱中保存,備用[9]。取樣品提取液1mL,加5%α-萘酚乙醇液3滴,搖勻后沿試管壁緩緩加入濃硫酸,觀察試管內液體顏色變化并記錄。
1.3 方法
1.3.1 色譜條件 Agilent Technologies 1220Infinity LC 高效液相色譜儀,Agilent HC-C18(2)色譜柱(4.6 mm×250 mm,5μm),以乙腈(A)和水(B)為流動相,采用梯度洗脫,梯度程序為:0 min:5% A;10 min:10% A;30 min:35% A;40 min:40% A;60 min:5% A。流 速1 mL/min,柱 溫30 ℃,檢測波長200nm,進樣量20μL[10]。
1.3.2 對照試驗的配置 精確稱取薯蕷皂苷元適量,加甲醇溶解配置成濃度為0.102 0mg/mL 的對照品溶液,0.45μm 的微孔濾膜過濾,備用。
1.3.3 高效液相圖譜供試品溶液制備 分別將干燥至恒重的藥材粉末2g和黃精顆粒精密稱取7g(相當于原藥材2g),置具塞錐形瓶中,按料液比為1:20加入80%的乙醇,超聲提取2次,溫度60℃,時間50min,過濾,合并濾液,蒸干,加入20mL 蒸餾水溶解,加入(20mL,20mL)的正丁醇萃取二次,取正丁醇層,旋干,加入少量甲醇溶解,分別置于10mL 的容量瓶中,加入甲醇定容,0.45μm 微孔濾膜過濾,取續濾液作為供試品。
1.3.4 精密度實驗 取黃精顆粒1號連續進樣5次進行測定,分別對共有峰保留時間和峰面積進行統計,計算RSD 值,結果發現它們的相對標準準偏差(RSD)均小于3%,因此,結果表明該儀器具有良好的精密度,見表1。

表1 精密度考察結果
1.3.5 穩定性實驗 將配好的薯蕷皂苷元標準品按照1.4.2含量測定的色譜條件分別上樣(8、10、12、14、16、18、20μL),以上樣量和峰面積做標準曲線見表2和圖1。 結果顯示:薯蕷皂苷元在0.816μg~2.04μg內線性良好,回歸方程:Y=20.662X+19.211,R2=0.999 4,R=0.999 7。
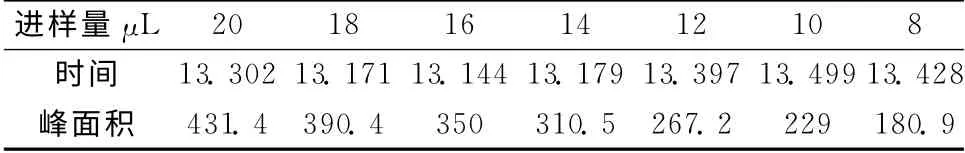
表2 薯蕷皂苷元的線性考察結果
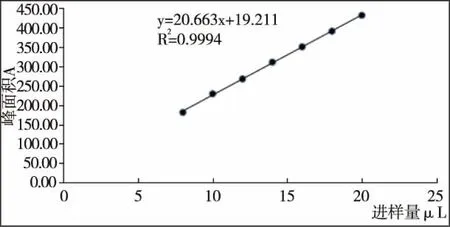
圖1 薯蕷皂苷元的線性關系
1.4.6 重現性實驗 取黃精顆粒3號供試品5份,按照1.3.2供試品溶液的制備方法和1.4.1色譜條件進行測定,統計各共有峰的保留時間和峰面積,計算RSD 值。結果如下圖表明它們的相對標準偏差(RSD)均都小于3%,因此表明該儀器的重現性良好,見表3。

表3 重復性考察結果
1.4.7 穩定性實驗 取黃精顆粒1 號供試品,在0、2、4、8、12小時依次進樣測定,統計各共有峰的保留時間和峰面積,計算各共峰的RSD 值。結果表明它們的相對標準偏差(RSD)均小于3%,因此說明該儀器在12小時內的穩定性良好,見表4。

表4 穩定性考察結果
2 結 果
經過與標準品對比可以看書5號峰是薯蕷皂苷元。通過顆粒和原藥材色譜圖對比得知顆粒和原藥材中的化學成分沒變化,黃精顆粒中薯蕷皂苷元的含量比原藥材中的薯蕷皂苷元含量高,見表5—7和圖2—4。

表5 黃精顆粒各峰的峰面積和保留時間

表6 原藥材各峰的峰面積和保留時間

表7 含量測定結果
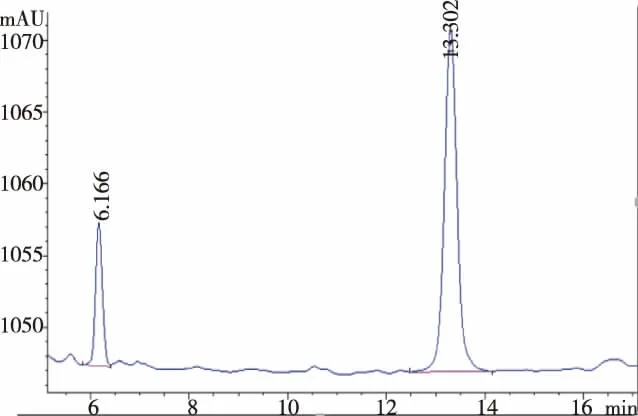
圖2 薯蕷皂苷元標準品色譜
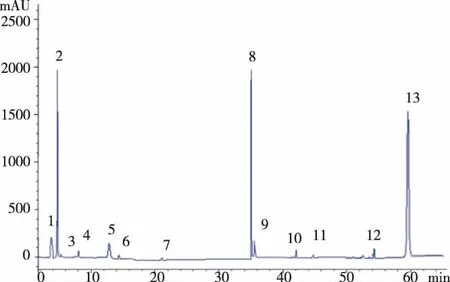
圖3 黃精原藥材色譜圖
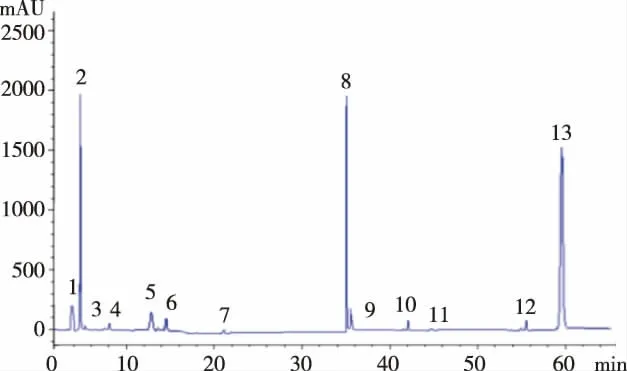
圖4 黃精顆粒的色譜圖
3 討 論
黃精顆粒是臨床上常用的中成藥物,具有滋補腎精、益氣補血功效,廣泛用于月經量少、后錯患者中[11-12]。但是,黃精顆粒臨床制備難度較大,本研究中黃精顆粒制備過程中多次均無法制備出顆粒,并且制備的顆粒均難以制成團,難以完成藥物過濾、篩選,經多次嘗試后發現,制備顆粒時需要在藥材浸潤成膏時給予蔗糖作為敷料,邊加邊攪拌成稠狀,然后在慢慢加入糊精制握之成團觸之即散的狀態[13]。同時,本研究中制劑、原料均給予超聲法提取,但是由于原材料中雜質相對較多,抽濾時可給予提取液通過濾紙極慢。因此,本研究中給予棉花將原藥材提取物中的雜質過濾一遍,然后再抽濾,這樣使得提取液更容易通過濾紙。此外,HPLC 制備時以甲醇、乙腈、水為流動相選擇最適合的流動相,通過實驗發現流動相以乙腈-水為佳。然后以乙腈-水為流動相采用二元梯度洗脫。通過不斷的更換流動相梯度,最后以1.4.1 高效液相圖譜的色譜條件中的梯度掃出的峰相對較多,峰型相對較好。因此選用1.4.1高效液相圖譜的色譜條件中的梯度進行實驗。
查閱文獻資料[14],選擇薯蕷皂苷元的檢測波長(200nm、210nm、203nm)進行測定,發現檢測波長在200nm 處出峰較號而且出峰較多。因此選用檢測波長為200nm 進行色譜研究。在方法學考察中對標準品線性進行考察時,剛開始掃出峰時間長而且雜峰多,還以為是標準品有問題,因此又換了不同廠家的標準品進行測定結果樣品不僅不出峰而且高效液相儀的梯度泵壓幾乎不到1,經過分析才發現可能是高效液相色譜儀器工作時間太長了,因此關了儀器等了幾天后使用儀器才恢復正常,標準品出峰的時間也比原來的出峰時間縮短了,而且峰性較好,峰面積穩定[15]。含量測定是制出的顆粒中薯蕷皂苷元的含量高于原藥材中薯蕷皂苷元的含量,可能是由于實驗存在正負誤差或者是由于顆粒在提取的時候比原藥材多提取了一次,因此做出這樣的結果,結果表明:經過與標準品對比可以看書5號峰是薯蕷皂苷元。通過顆粒和原藥材色譜圖對比得知顆粒和原藥材中的化學成分沒變化,黃精顆粒中薯蕷皂苷元的含量比原藥材中的薯蕷皂苷元含量高。
綜上所述,將HPLC法用于黃精顆粒和提取物含量測定中效果理想,具有準確、簡便,重現性好等優點;黃精顆粒中的薯蕷皂苷元含量比原藥材中的薯蕷皂苷元含量高。
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
當代陜西(2019年8期)2019-05-09 02:22:48
上海建材(2019年1期)2019-04-25 06:30:48
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
家庭影院技術(2018年4期)2018-05-09 07:07:52
專用汽車(2016年4期)2016-03-01 04:13:43
質量與標準化(2015年9期)2015-12-31 11:41:40
中國質量與標準導報(2014年4期)2014-03-11 19:54:25
中國質量與標準導報(2014年10期)2014-02-28 22:25:47
中國質量與標準導報(2014年7期)2014-02-28 22:24:39
