范可尼貧血FANCA基因雙重雜合突變1例報告并基因突變分析
2018-11-13 01:25:56楊婭平李棟梁曹瑋趙田華
山東醫藥 2018年38期
楊婭平,李棟梁,曹瑋,趙田華
(1承德醫學院,河北承德067000;2解放軍白求恩國際和平醫院)
范可尼貧血(FA)是一種罕見的常染色體或X染色體連鎖的隱性遺傳病,臨床表現為進行性加重的造血功能衰竭、先天性畸形及高風險進展為急性髓系白血病[2],約50%的患者呈原發不孕[3,4]。FA的發生源于FA基因突變。FA基因是一組在DNA交聯損傷中起同源重組修復(HRR)作用的基因[1],其常見的突變類型包括FANCA、FANCC和FANCG。目前認為,FA是由于DNA內切酶異常阻礙了受損DNA自然修復過程,進而引起染色體畸變,最終導致造血干細胞缺陷及多發性畸形。常規診斷FA主要依據全血細胞減少家族史、骨髓再生障礙、特征性先天畸形及染色體斷裂等。目標序列捕獲高通量測序是近年發展起來的一項新的測序技術,能夠同時篩查多種基因突變。我們應用此技術檢出1例FA患兒攜帶FANCA基因雙重突變,用Sanger測序法加以驗證并檢測其父母、姐姐基因型。現報告如下。
1 資料與方法
1.1 臨床資料 患兒女,9歲,因“間斷乏力、鼻衄1年10月余”,于2015年11月6日來我院就診。患兒既往在當地醫院依據血常規、骨髓穿刺結果考慮再生障礙性貧血(具體檢查結果未知),日常間斷口服康力龍及環孢素治療,療效不滿意。患兒父母非近親結婚,家族中無類似疾病史。查體:發育落后于同齡兒,無骨骼畸形,中度貧血貌,皮膚未見色素沉著及出血點,心臟未聞及異常雜音,肝脾肋下未觸及腫大。實驗室檢查:①血常規:白細胞3.0×109/L,中性粒細胞36.5%,淋巴細胞60.8%,紅細胞2.6×1012/L,血紅蛋白102.0 g/L,血小板38.0×109/L。②骨髓象:有核細胞增生減低,粒系比例略低,占26.5%,均為中幼及以下階段,胞質顆粒明顯增多;紅系比例明顯增高,占57.5%,以中晚幼紅細胞為主,偶見雙核紅細胞,成熟紅細胞大小不一;淋巴細胞比例基本正常;全片共見巨核細胞4個,血小板散在少見。③骨髓活檢:骨髓增生極度低下(10%),伴明顯出血,少量粒、紅系細胞散在分布,分葉核為主,巨核細胞可見,偶見胞體小的巨核細胞。網狀纖維染色(MF)0級。免疫組化:CD34、CD14、CD117陰性,CD61、CD42b、MPO陽性,CD68少數陽性。④骨髓增生異常綜合征(MDS)及白血病相關基因檢測:骨髓細胞FISH檢查MDS、白血病相關基因EGR1、CEP7、TP53、CEP8均未見異常;Sanger測序示MDS、白血病相關基因IDH1、IDH2、NPM1、DNMT3A、SRSF2、SF3B1及U2AF1均未見異常。④染色體分析:核型為46,XX;染色體斷裂實驗未見異常。⑤腎臟B超:左腎缺如,右腎融合腎。考慮患兒年幼伴發先天畸形,發育遲緩,間斷口服康力龍及環孢素癥狀緩解差,可能為骨髓衰竭綜合征,尤以FA為著,因此做FA相關檢查。以50例健康志愿者作為對照排除基因多態性。本研究經醫院倫理委員會批準,并征得患兒及家屬知情同意。
1.2 患兒FANCA突變基因檢測及驗證
1.2.1 基因組DNA提取 采集患兒外周血3~5 mL,EDTA抗凝,按DNA提取試劑盒說明書提取外周血DNA,-20 ℃保存備用。
1.2.2 FANCA突變基因檢測 采用目標序列捕獲和高通量測序技術。利用基于第二代測序方法設計的包含249種先天性骨髓衰竭癥的診斷試劑盒,首先將提取的基因組DNA用超聲波打斷并制備DNA文庫,然后通過捕獲芯片對FANCA基因編碼區及其側翼序列的DNA進行富集,用通用引物對捕獲到的序列進行PCR擴增,最后應用Illumina Hiseq測序平臺對擴增產物進行高通量測序,尋找突變的致病基因。
1.2.3 FANCA突變基因鑒定 采用Sanger測序技術。在確定患兒突變的致病基因后,對患兒外周血DNA進行測序驗證。引物設計采用Primer Premier5.0軟件,由上海生工生物合成。PCR擴增反應體系(50 μL):DNA模板5 μL,10×PCR緩沖液5 μL,l×dNTP 5 μL,Taq酶2 U,上、下游引物各1.5 μL,補雙蒸水至50 μL。反應條件:94 ℃ 30 s,55~65 ℃ 40 s,72 ℃ 45 s,共35個循環。將PCR產物在瓊脂糖凝膠上進行電泳,紫外燈下觀察。DNA測序:PCR產物純化后,用測序試劑盒在ABI3130 DNA測序儀上進行序列測定。將檢測結果與人類基因突變數據庫(HGMD)進行比對,確定有無此類突變的報道。另外對50例健康人進行該基因突變的篩查作為陰性對照。
1.3 患兒家系直系成員FANCA突變基因檢測 采集患兒父母、姐姐的外周血,采用Sanger測序技術檢測外周血DNA的基因型。方法同上。
1.4 突變危害性的預測 應用Mutation Taster和PolyPhen-2軟件預測基因突變對蛋白功能的影響程度。當兩種軟件均預測同一突變對蛋白的功能影響較大時,認定該突變具有較強的危害性。
2 結果
2.1 患兒FANCA突變基因檢測結果 目標序列捕獲測序分析FANCA基因上所有外顯子的測序覆蓋率均達到99%以上,各外顯子上的平均測序深度和測序深度中位數均比較接近,提示測序的隨機性比較好。通過目標序列捕獲高通量測序技術檢測患兒FANCA基因的所有外顯子及其側翼序列,發現16號染色體上FANCA基因編碼區存在雙重雜合突變,即第14外顯子c.1303C>T(p.R435C)和第31外顯子c.3031C>T(p.R1011C),導致該基因的第435位和1011位氨基酸均由精氨酸變成半胱氨酸。見圖1、2。
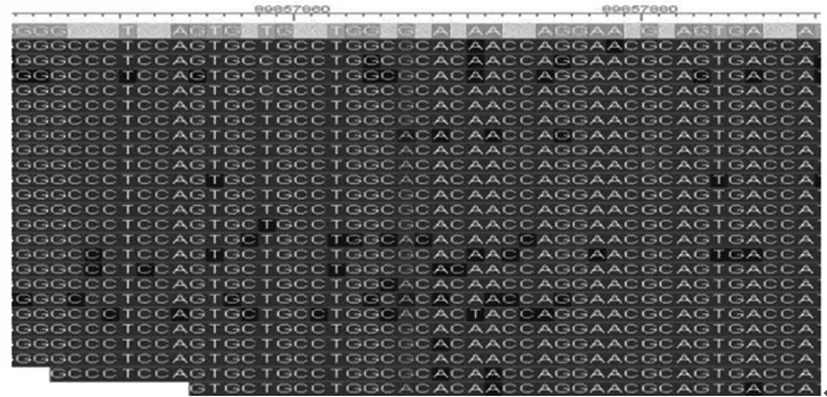
圖1 患兒FANCA基因c.1303C>T突變目標序列捕獲和高通量測序圖
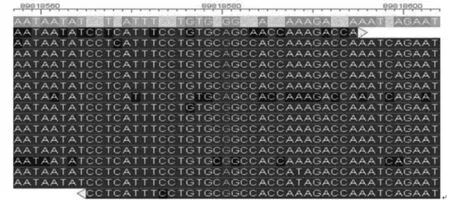
圖2 患兒FANCA基因c.3031C>T突變目標序列捕獲和高通量測序圖
2.2 患兒FANCA突變基因驗證結果 應用Sanger測序法對先證者的上述突變位點進行驗證,其結果與目標序列捕獲高通量測序法完全一致。見圖3、4。對50例健康個體FANCA基因相應區域測序,均未發現上述序列改變。
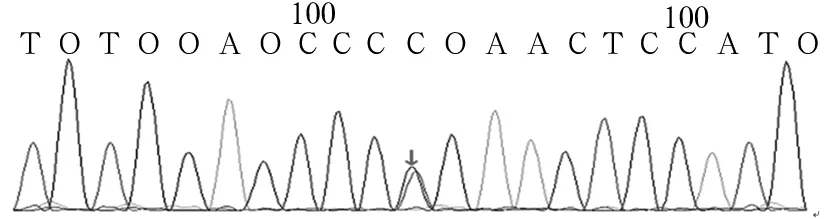
圖3 患兒FANCA基因c.1303C>T突變Sanger測序圖
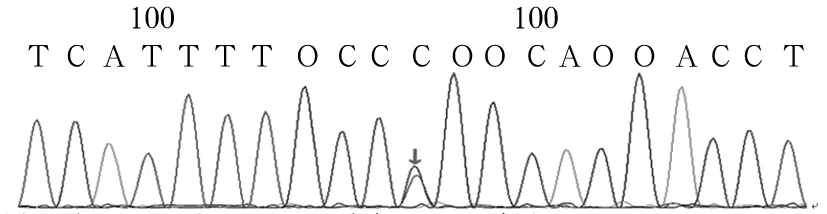
圖4 患兒FANCA基因c.3031C>T突變Sanger測序圖
2.3 患兒家系直系成員FANCA突變基因檢測結果 父親攜帶第31外顯子c.3031C>T(p.R1011C)突變,母親及姐姐均攜帶第14外顯子c.1303C>T(p.R435C)突變。
2.4 蛋白功能預測分析結果 c.3031C>T(p. R1011C)和c.1303C>T(p.R435C)對蛋白功能影響均為有害。
3 討論
多數FA患者出生時血細胞正常或接近正常,在5~10歲時出現血細胞減少[5],常合并貧血、出血和感染,部分患者表現為進行性骨髓衰竭與繼發性腫瘤如急性髓系白血病等,造血功能衰竭合并感染為該病常見的死亡原因。隨著年齡增長,FA患者可出現多發畸形與發育遲滯,其中畸形的發生率約為60%[6]。FA的實驗室檢查包括血象、骨髓象及細胞遺傳學檢測等。其血液學改變程度和類型呈高度異質性;骨髓象類似于獲得性再障,表現為增生減低、造血細胞減少、非造血細胞增多;細胞遺傳學檢測可見染色體斷裂、缺失,染色單體互換、核內再復制、環形染色體畸變等不穩定現象。Auerbach等[7]報道了222例FA患者,其中44%表現為骨髓造血異常和先天畸形,51%僅有造血異常或先天畸形,5%兩者皆無。但也有再障患兒伴先天畸形而無細胞遺傳學改變者,因此僅靠臨床表現可能漏診或誤診。本例患兒自幼身材矮小,發育遲于同齡正常兒童,7歲左右出現乏力、鼻衄癥狀,血常規提示全血細胞減少,骨髓象提示骨髓造血功能低下,超聲發現左腎缺如及右側融合腎,符合FA患者發育遲緩、骨髓造血衰竭及先天畸形的臨床特點。
近年來,利用體細胞融合雜交技術和FA細胞對DNA交鏈劑異常敏感的基因缺陷進行互補分析,目前已發現有19種基因被證實參與FA蛋白的相關功能。這些基因以“FANC”為詞根進行命名,FANCA、FANCC和FANCG是3種較為常見的基因突變,約占FA患者的85%,其中FANCA基因亞型最為常見[8]。FANCA約有200個不同的等位基因,包括幾乎所有的已知的突變類型,大片段的缺失是主要的突變類型。轉化為骨髓增生異常綜合征及白血病的FA通常有基因或染色體異常,其中7號和3q異常的預后差[9,10]。
目標序列捕獲高通量測序是將目標序列捕獲與高通量測序相整合進行DNA序列分析的新技術,具有速度快、準確率高、成本低、覆蓋度廣以及與生物學表型結合更為直接等優點,能夠同時檢測大量基因,適用于遺傳異質性疾病的基因突變篩查,具有很好的應用潛力,亦為檢測FA相關的基因突變提供了更為準確和直觀的診斷依據。本研究應用該技術對1例疑似FA患兒進行了249種骨髓衰竭癥相關基因突變的篩查,發現本例患兒存在16號染色體上FANCA基因第14外顯子c.1303C>T和第31外顯子c.3031C>T雙重雜合突變,并用Sanger測序得以驗證,從而確定了FA的診斷。有關FANCA基因14外顯子c.1303C>T突變國內外僅見1例報道,即在97例FA患者中檢測到40種致病基因突變位點,其中1例為c.1303C>T突變,且此突變與FA發病密切相關[11];后一突變位點通過查詢千人基因組計劃數據庫(HGMD)和美國國家生物技術信息中心單核苷酸多態性數據庫(dbSNP)目前尚未見報道。我們進一步應用蛋白功能預測軟件檢索提示,上述2個突變對蛋白功能影響均為有害。
FA的治療首選異基因造血干細胞移植,可修復受損細胞及改善FA造血微環境,是提高患者長期無病生存率的惟一方法;無移植條件者可給予雄激素、細胞因子如粒細胞集落刺激因子及對癥支持治療。該患兒平時間斷口服雄激素及輸血治療,目前病情穩定,正在進一步隨訪中。
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學生導刊(2018年13期)2018-06-29 03:49:00
現代檢驗醫學雜志(2016年4期)2016-11-15 02:01:14
海峽科技與產業(2016年3期)2016-05-17 04:32:12
