603樹脂體系及其熱熔預(yù)浸料室溫儲存期性能研究*
2018-07-20 00:26:02樊孟金白雪蓮石佩洛潘玲英
固體火箭技術(shù) 2018年3期
關(guān)鍵詞:復(fù)合材料
臧 千,樊孟金,白雪蓮,周 宇,石佩洛,潘玲英
(航天材料及工藝研究所,北京 100076)
0 引言
纖維增強(qiáng)樹脂基復(fù)合材料是以聚合物樹脂作為基體、纖維為增強(qiáng)材料的典型先進(jìn)復(fù)合材料,具有高比強(qiáng)度、高比模量、耐高溫、低密度、可設(shè)計(jì)性好、抗疲勞性能及減震性能好等一系列的優(yōu)異性能,能制成大型承力結(jié)構(gòu)件,廣泛用于航空航天和民用工業(yè)領(lǐng)域[1-3]。
預(yù)浸料是制備纖維增強(qiáng)樹脂基復(fù)合材料的重要中間材料,是復(fù)合材料性能的基礎(chǔ)。復(fù)合材料成型時的工藝性能和力學(xué)性能取決于預(yù)浸料的性能。當(dāng)前,國外實(shí)現(xiàn)成熟應(yīng)用的炭纖維樹脂基結(jié)構(gòu)復(fù)合材料和預(yù)浸料是以日本東麗T800和美國赫氏IM7為代表的高強(qiáng)中模炭纖維增強(qiáng)/高韌性樹脂作為基體的第二代先進(jìn)復(fù)合材料和預(yù)浸料。國內(nèi)多個廠家的T800級別炭纖維已經(jīng)逐漸形成了百噸級批量制備能力和工程應(yīng)用的推廣。近年來,國內(nèi)外通過超聲波將炭纖維預(yù)浸料層板擴(kuò)展薄層化后,使得單層纖維預(yù)浸料的厚度更薄(低至0.04 mm),其他預(yù)浸料技術(shù)的新進(jìn)展包括可常溫儲存的預(yù)浸料,低溫固化高溫使用的預(yù)浸料,大絲束炭纖維預(yù)浸料和快速固化預(yù)浸料等。值得注意的是,預(yù)浸料應(yīng)具有適當(dāng)?shù)牟僮鞴に囆裕凑承院弯伕残浴U承允軠囟群蛢Υ嫫诘挠绊戄^大,同一片預(yù)浸料,溫度低可能失去粘性,溫度高又有粘性,另外隨著儲存期的延長,預(yù)浸料中樹脂預(yù)聚度發(fā)生一定變化,從而會影響預(yù)浸料的鋪覆工藝性和后期復(fù)合材料的性能。遺憾的是,粘性的評價目前還沒有找到一個非常適宜的客觀方法。文獻(xiàn)中報道的預(yù)浸料粘性的研究方法包括垂直測試法[4]、滾球法[5]和楔子剝離法[5]等。也有通過樹脂動態(tài)復(fù)數(shù)粘度(η*)和玻璃化轉(zhuǎn)變溫度(Tg)來表征預(yù)浸料粘性的相關(guān)研究[6]。一些航天器等大型復(fù)合材料結(jié)構(gòu)件由于預(yù)浸料的用量較多,且其生產(chǎn)周期較長,對預(yù)浸料的質(zhì)量及工藝性要求較高,進(jìn)而對其室溫儲存期的過程監(jiān)控提出了更高要求。針對預(yù)浸料儲存期的研究還不夠深入,目前主要集中在對樹脂凝膠時間[7]、流變特性[8]等物理性能和復(fù)合材料力學(xué)性能[7]的影響等方面,缺少對固化度的評價表征。
本文針對一種耐高溫環(huán)氧樹脂預(yù)浸料TGT800-12K/603,考察室溫儲存期對樹脂及其預(yù)浸料各項(xiàng)性能的影響,以確定該樹脂及預(yù)浸料的室溫儲存期及室溫儲存過程中固化度評價方法,為其在大型復(fù)合材料結(jié)構(gòu)件上的使用周期提供理論依據(jù),并為其推廣應(yīng)用提供理論參考。
1 實(shí)驗(yàn)
1.1 原材料
熱熔預(yù)浸料用耐高溫環(huán)氧樹脂體系,603,航天材料及工藝研究所。
炭纖維,TGT800-12K,山西鋼科碳材料有限公司。
1.2 實(shí)驗(yàn)過程
預(yù)浸料制備:將TGT800-12K炭纖維分別與603樹脂體系復(fù)合,制成面密度165 g/m2,樹脂質(zhì)量含量34%的C/E預(yù)浸料,預(yù)浸料牌號為TGT800-12K/603。
預(yù)浸料儲存:模擬大型復(fù)合材料結(jié)構(gòu)件的生產(chǎn)過程,將聚乙烯膜從預(yù)浸料表面揭開,置于室溫條件下,定期取出,進(jìn)行實(shí)驗(yàn)。
1.3 表征與測試
樹脂粘度測試,采用DV2旋轉(zhuǎn)粘度儀(美國博勒飛粘度儀公司),升溫速率1 ℃/min;樹脂低溫下玻璃化轉(zhuǎn)變溫度及固化反應(yīng)特性分析,采用差示掃描熱分析儀(DSC)(美國梅特勒-托利多公司),升溫速率10 ℃/min;樹脂凝膠時間測試,采用凝膠時間測定儀(北京化學(xué)試劑公司)按照GB/T12007.7—1989,測試溫度180 ℃;預(yù)浸料樹脂含量、揮發(fā)物含量測試,采用DGF313P型可編程電熱鼓風(fēng)干燥箱(重慶顥園環(huán)境實(shí)驗(yàn)設(shè)備有限公司),按照J(rèn)C/T776—2004、JC/T780—2004方法,試樣尺寸為100 mm×100 mm;樹脂流動度測試,采用千斤頂壓機(jī)(航利程序控制有限公司),按照J(rèn)C/T775—2004方法,試樣尺寸為50 mm×50 mm,鋪層順序?yàn)閇0/90]。
2 結(jié)果與討論
2.1 樹脂室溫儲存期性能
2.1.1 樹脂粘度和凝膠時間
預(yù)浸料粘性和樹脂流動度發(fā)生變化的本質(zhì)是樹脂發(fā)生固化反應(yīng),使分子鏈延長或產(chǎn)生微凝膠,導(dǎo)致樹脂粘度增大,軟化點(diǎn)升高。圖1為不同室溫儲存時間的603環(huán)氧樹脂等速升溫的粘度曲線,測試升溫速率為1 ℃/min。由圖1可看出,樹脂最低粘度出現(xiàn)在150 ℃左右,最低粘度值由最初的2.240 Pa·s,到35 d后上升到9.478 Pa·s,是其初始粘度值的4.2倍。說明隨著室溫儲存時間延長,環(huán)氧樹脂在室溫下發(fā)生反應(yīng),樹脂分子量增大,表現(xiàn)出粘度增大的趨勢。
測試了不同室溫儲存時間的603環(huán)氧樹脂180 ℃下凝膠時間。如表1所示,603樹脂在180 ℃下凝膠時間由最初的25.17 min,到35 d后變成16.49 min,凝膠時間顯著減小。
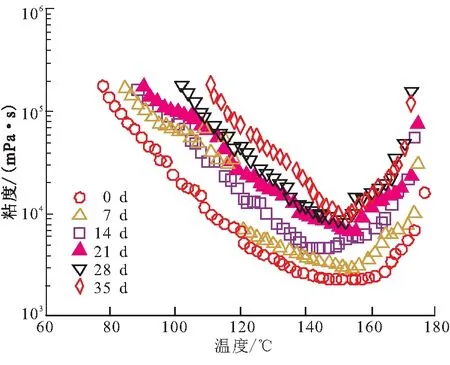
圖1 不同儲存時間603環(huán)氧樹脂的粘度曲線Fig.1 Viscosity vs temperature curve of 603 resin at different shelf life

表1 不同儲存時間603環(huán)氧樹脂180 ℃下凝膠時間Table1 Gelation time at 180 ℃ of 603 resin at different storage life
2.1.2 未固化樹脂玻璃化轉(zhuǎn)變溫度及固化反應(yīng)特性
采用DSC分析儀對未固化603樹脂玻璃化轉(zhuǎn)變溫度、固化反應(yīng)特性進(jìn)行表征,如圖2所示。可以看出,DSC曲線表明,樹脂未固化的玻璃化轉(zhuǎn)變溫度比較明顯,出現(xiàn)在-5.3 ℃。隨著測試溫度的升高,在278 ℃出現(xiàn)熱流峰值(Tp)。
一般取DSC曲線熱流q斜率最大的點(diǎn)為Tg。采用式(1)對DSC曲線進(jìn)行微分,微分曲線峰值位置即為Tg。前期研究結(jié)果表明,通過測量未固化樹脂Tg可以實(shí)現(xiàn)對于C/E預(yù)浸料的粘性這一主觀術(shù)語的客觀表征,Tg=0 ℃這個區(qū)域附近的環(huán)氧樹脂體系制備出的C/E預(yù)浸料具有最佳的操作工藝性。這是由于Tg值越高,樹脂室溫粘度越大,預(yù)浸料的操作工藝感受越“干”[6]。
(1)
圖3表示603樹脂Tg隨著儲存時間的變化。結(jié)果表明,603樹脂在室溫儲存條件下緩慢反應(yīng)時,隨著時間的推移,其Tg由初始值-5.3 ℃,逐漸增加,35 d后變成4.3 ℃,累計(jì)增加了9.6 ℃。說明隨著儲存期變化,樹脂發(fā)生緩慢的固化反應(yīng),分子量逐漸增大,因此Tg逐漸升高。
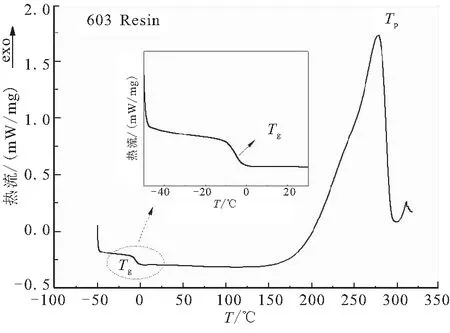
圖2 603樹脂體系的DSC曲線Fig.2 DSC curve of 603 resin system

圖3 不同儲存時間603環(huán)氧樹脂的Tg變化圖Fig.3 Change of Tg of 603 resin at different storage life
圖4表示603樹脂Tp隨著儲存期的變化。結(jié)果表明,隨著儲存期時間的變化,樹脂反應(yīng)峰值溫度(Tp)逐漸降低,由初始峰值278.2 ℃下降到35 d后的275.7 ℃,累計(jì)下降了2.5 ℃,變化較小。
取DSC曲線熱流q的積分面積(ΔH)表示樹脂固化反應(yīng)的放熱量:

(2)
定義樹脂初始狀態(tài)的樹脂固化反應(yīng)的放熱量為ΔH0,則樹脂的固化度α可以通過式(3)計(jì)算:
(3)
圖5表示603樹脂ΔH和α隨著儲存期的變化。結(jié)果表明,樹脂固化反應(yīng)的放熱量ΔH逐漸減少,樹脂固化度α逐漸增加。室溫儲存35 d后,已經(jīng)產(chǎn)生了11%的固化度。

圖4 不同儲存時間603環(huán)氧樹脂的Tp變化圖Fig.4 Change of Tp of 603 resin at different storage life

圖5 不同儲存時間603環(huán)氧樹脂的ΔH和α變化圖Fig.5 Change of ΔH and α of 603 resin at different shelf life
2.2 熱熔預(yù)浸料室溫儲存期性能
2.2.1 揮發(fā)物含量
圖6為不同儲存時間熱熔預(yù)浸料的揮發(fā)物含量。從圖6可以看出,隨著儲存時間的延長,預(yù)浸料揮發(fā)物含量逐漸變大,由最初的0.3%上升到1.2%。由于采用了無溶劑的熱熔預(yù)浸料制備方法,因此預(yù)浸料初始揮發(fā)份含量較低(0.3%)。覆蓋聚乙烯膜的預(yù)浸料,揮發(fā)份含量一般保持不變[7-8]。該試驗(yàn)儲存條件為揭開預(yù)浸料表面的聚乙烯膜,因此預(yù)浸料在室溫環(huán)境的影響下吸濕引起了揮發(fā)份含量增加。
2.2.2 預(yù)浸料樹脂流動度
樹脂的流變性能對復(fù)合材料的成型工藝具有決定性的影響,而樹脂流動度反映了預(yù)浸料的流變性能。因此可用來評價預(yù)浸料的儲存[9-11]。圖7為熱熔預(yù)浸料儲存期間樹脂流動度測試數(shù)據(jù),如數(shù)據(jù)顯示,隨著儲存時間的延長和樹脂的固化程度增加,流變性能變差,35 d后,樹脂流動度由初始的20%下降到14%。一般認(rèn)為樹脂流動度需大于15%才能保證適當(dāng)?shù)某尚凸に囆裕虼藘Υ?5 d的預(yù)浸料已經(jīng)不利于鋪覆和復(fù)合材料結(jié)構(gòu)件的成型,這與圖1樹脂粘度的變化規(guī)律是相一致的,即樹脂粘度增加,流動度降低。

圖6 不同儲存時間熱熔預(yù)浸料的揮發(fā)物含量Fig.6 Volatiles content of prepregs at different shelf life

圖7 不同儲存時間熱熔預(yù)浸料的樹脂流動度Fig.7 Resin flow of prepregs at different storage life
2.2.3 預(yù)浸料粘性
表2為熱熔預(yù)浸料儲存期間的粘性變化。結(jié)果表明,“新鮮”的預(yù)浸料粘性好,超過35 d的預(yù)浸料已經(jīng)完全失去粘性發(fā)干,無法鋪貼。這是因?yàn)殡S著樹脂固化度的提高,樹脂從半固態(tài)變成固態(tài),預(yù)浸料失去柔軟性,沒有加熱輔助,已不能將預(yù)浸料鋪在一起,說明樹脂體系已達(dá)到相當(dāng)固化程度,使其軟化點(diǎn)高于室溫[12]。

表2 不同儲存時間熱熔預(yù)浸料的粘性Table2 Tack properties of prepregs at different shelf life
2.3 討論
以上603樹脂和TGT800-12K/603預(yù)浸料的各項(xiàng)測試結(jié)果存在一致性關(guān)聯(lián),均表明隨著儲存期的延長,樹脂固化度提高,預(yù)浸料的操作工藝性下降。
在實(shí)際工程應(yīng)用中,針對預(yù)浸料發(fā)“干”鋪覆性下降的狀態(tài),一般進(jìn)行DSC測試通過反應(yīng)放熱量ΔH推算出樹脂的固化度。由于樹脂難以從預(yù)浸料中分離出來,因此DSC測試對象是含有纖維的預(yù)浸料,需進(jìn)一步通過預(yù)浸料中樹脂含量進(jìn)行推算,過程見式(4):
(4)
式中 ΔHprepreg為預(yù)浸料試樣DSC放熱量測試值;w為預(yù)浸料中樹脂含量,本實(shí)驗(yàn)取w=34%。
由于DSC測試試樣較小(僅為約10 mg),針對該試樣的w值和宏觀測試值可能存在較大偏差,所以該計(jì)算方法推算出的固化度也可能存在較大偏差。本實(shí)驗(yàn)的結(jié)果表明,603樹脂的α和Tg存在相關(guān)性,見圖8。因此,通過測量包含部分603樹脂的TGT800-12K/603預(yù)浸料試樣的Tg,即可以通過圖8直接判斷預(yù)浸料中樹脂的α。該方法效率更高,結(jié)果更可靠。
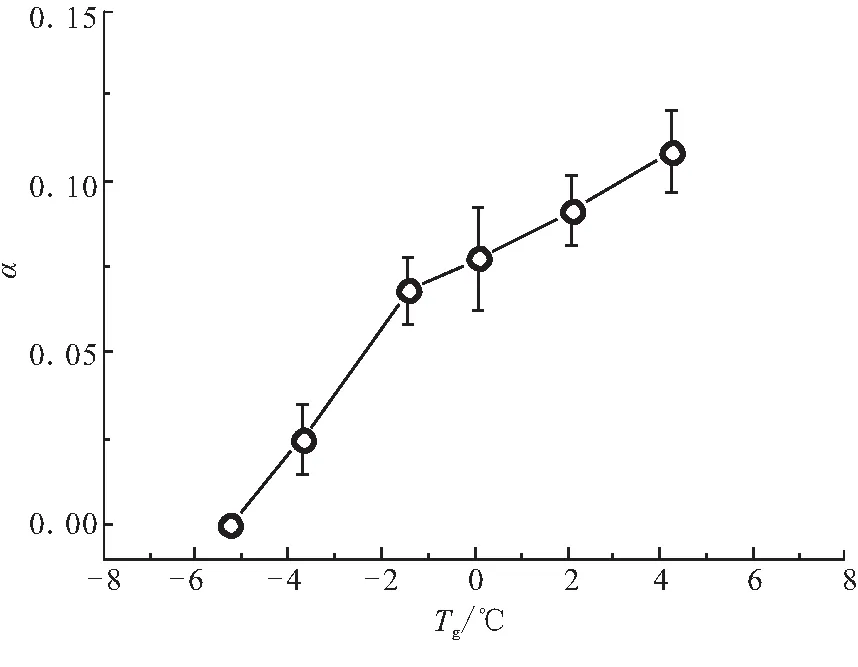
圖8 603樹脂的α和Tg關(guān)系圖Fig.8 Relationship of α vs Tg of 603 resin
3 結(jié)論
(1)室溫儲存35 d后,603樹脂最低粘度上升4.2倍,Tg增加了9.6 ℃,固化度為11%;TGT800-12K/603預(yù)浸料樹脂流動度由初始的20%下降到14%,粘性變差,失去操作工藝性。
(2)TGT800-12K/603預(yù)浸料樹脂的固化度α可以通過DSC測試預(yù)浸料的Tg來進(jìn)行高效準(zhǔn)確的評估。
猜你喜歡
建材發(fā)展導(dǎo)向(2022年2期)2022-03-08 01:44:04
建材發(fā)展導(dǎo)向(2021年14期)2021-08-23 00:56:16
中國材料進(jìn)展(2019年10期)2019-12-07 05:32:14
纖維復(fù)合材料(2018年3期)2018-04-25 07:22:58
電子測試(2017年11期)2017-12-15 08:57:13
山東工業(yè)技術(shù)(2016年15期)2016-12-01 05:31:34
中國塑料(2015年6期)2015-11-13 03:02:54
中國塑料(2015年11期)2015-10-14 01:14:14
中國塑料(2015年8期)2015-10-14 01:10:41
應(yīng)用化工(2014年10期)2014-08-16 13:11:29
