丹酚酸A對脂多糖誘導人肺微血管內皮細胞線粒體自噬和細胞損傷的影響
2018-05-11 00:02:40李偉文藍秀黎媛孫蕾呂祝慶
溫州醫科大學學報 2018年4期
關鍵詞:檢測
李偉文,藍秀,黎媛,孫蕾,呂祝慶
(溫州醫科大學附屬第五醫院 呼吸內科,浙江 麗水 323000)
人肺微血管內皮細胞(human pulmonary microvascular endothelial cells,HPMVECs)是構成肺泡-毛細血管屏障的重要成分。在肺炎癥性疾病中,內毒素誘導的HPMVECs損傷是導致肺泡-毛細血管屏障破壞,最終導致急性肺損傷,甚至導致急性呼吸窘迫綜合征的重要原因之一[1-2],預防和治療HPMVECs損傷是降低患者病死率的重要手段[2-3]。丹酚酸A(salvi-anolic acid A,Sal A)是從丹參根莖中提取的重要活性成分,具有清除氧自由基、降低細胞內氧化應激、減少細胞損傷的作用[4-5]。本研究以脂多糖(lipopolysaccharides,LPS)作為誘導劑,構建HPMVECs損傷模型,觀察Sal A對LPS誘導的HPMVECs損傷的保護作用,探討其可能的作用機制。
1 材料和方法
1.1 材料 HPMVECs購于美國ScienCell公司,Sal A、MTT、RPMI 1640培養基購于美國Sigma公司,10%胎牛血清購于美國Gibco公司,ROS檢測試劑盒和JC1試劑盒購自上海碧云天生物技術有限公司,線粒體蛋白提取試劑盒購于上海貝博生物科技有限公司,Beclin1、LC3 II/I、PINK1、Parkin和Tubulin單克隆抗體購于美國CST公司。
1.2 方法
1.2.1 細胞培養:HPMVECs用含鏈霉素、氯霉素和10%胎牛血清的RPMI 1640培養基傳代,培養于37 ℃、5% CO2、飽和濕度的細胞培養箱中。細胞融合達80%以上時傳代,取第4~第8代細胞進行實驗。
1.2.2 細胞增殖檢測:按照文獻[6]方法,以細胞密度為2×104/mL接種于96孔板,每孔200 μL,次日換無血清培養基。用0.5、2.5、5、10、25、50 μmol/L Sal A處理48 h;分為4組:對照組、Sal A組、LPS組、LPS+Sal A組,Sal A組用終濃度為50 μmol/L的Sal A處理,LPS組用終濃度為10 μg/mL LPS處理,LPS+Sal A組用10 μg/mL的LPS和50 μmol/L的Sal A共培養,對照組不加藥,并設置空白對照為調零孔,每組設置6個復孔,繼續培養24 h、48 h和72 h。經上述處理后,每孔加入20 μL MTT試劑,加入二甲基亞砜后,用酶聯免疫檢測儀在492 nm波長處測各孔的吸光度值(A值)。細胞相對存活率=(受試組A值-空白對照A值)/(對照組A值-空白對照A值)×100%。
1.2.3 ROS相對含量和線粒體膜電位檢測:按上述分組,繼續培養48 h后,按照ROS試劑盒檢測方法,用終濃度為10 μmol/L的DCFH-CA重懸細胞,置入37 ℃細胞培養箱內孵育20 min,以無血清細胞培養液洗滌3次后,用流式細胞技術分析細胞內ROS含量改變。按照JC1試劑盒說明書操作方法收集1×106個細胞,用JC1染色緩沖液洗滌2次,加入10 μg/mL的JC1試劑,置入37 ℃細胞培養箱內孵育20 min,用流式細胞儀檢測紅色熒光強度改變。以對照物為標準,計算各組ROS和JC1的相對含量。
1.2.4 蛋白提取及濃度測定:按上述分組繼續培養48 h后,棄上清,用預冷PBS漂洗2次,加入120~150 μL RIPA裂解液冰上裂解10~15 min,4 ℃離心機12000 r/min離心15 min,取上清,即為細胞總蛋白。根據線粒體提取試劑盒說明書,將各組細胞培養48 h后,收集細胞,加入200 μL試劑A,冰上靜置10 min,勻漿后,離心,取上清液,11000×g離心后,棄上清,加入試劑B,再次離心后,取沉淀物,加入100 μL的裂解液,收集線粒體蛋白。采用BCA法檢測細胞總蛋白和線粒體蛋白濃度。
1.2.5 免疫印跡法檢測細胞及線粒體自噬蛋白改變:取20 μg細胞總蛋白或者線粒體蛋白4%~12%聚丙烯酰胺梯度凝膠電泳分離,電轉至PVDF膜,3%脫脂牛奶室溫封閉30~60 min。加入一抗(Beclin1、LC3 II/I、PINK1、Parkin和Tubulin),4 ℃孵育過夜;PBST緩沖液洗滌3次,每次8~10 min,加入HRP標記二抗,室溫孵育1~2 h,PBST緩沖液洗膜3次,每次8~10 min,用ECL試劑發光、顯影和定影,Image J軟件進行蛋白顯帶顏色深淺和面積計算,以Tubulin為內參。上述實驗重復3次。
1.3 統計學處理方法 采用SPSS22.0統計軟件進行分析。計量資料以±s表示,采用單因素方差分析和重復測量資料方差分析。P<0.05為差異有統計學意義。
2 結果
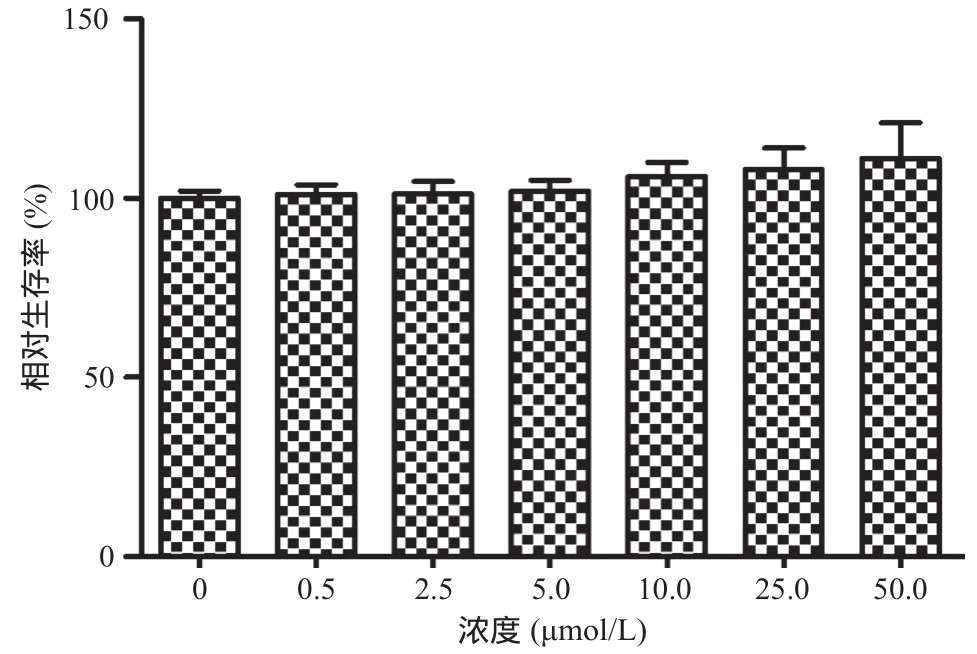
圖1 Sal A對HPMVECs相對存活率的影響
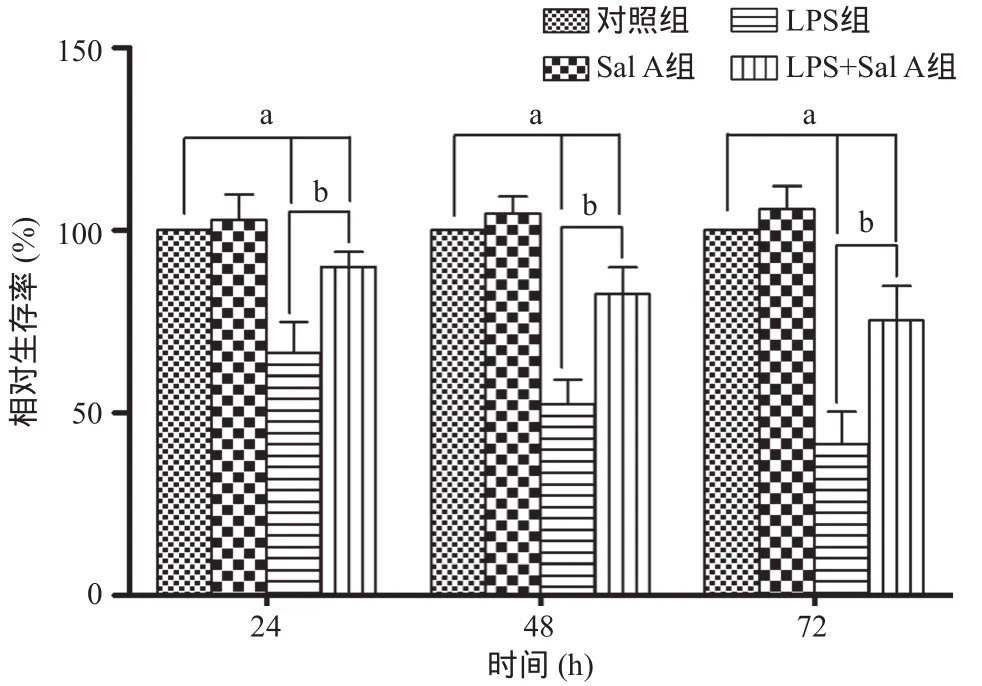
圖2 Sal A對LPS誘導HPMVECs損傷的影響
2.1 Sal A抑制LPS誘導的細胞損傷作用 圖1顯示以終濃度為0.5、2.5、5、10、25、50 μmol/L的Sal A處理HPMVECs 48 h后,細胞增殖無顯著抑制,與對照組比差異無統計學意義(P>0.05)。圖2顯示,50 μmol/L Sal A處理24 h、48 h和72 h后,細胞相對存活率無明顯降低,與對照組比差異無統計學意義(P>0.05)。10 μg/mL LPS處理24 h、48 h和72 h后,細胞相對存活率降低;與LPS組比,LPS+Sal A組24 h、48 h和72 h時細胞相對存活率顯著增高(P<0.05),但低于對照組(P<0.05)。
2.2 Sal A對LPS誘導HPMVECs的ROS含量和線粒體膜電位的影響 ROS活性檢測結果顯示:LPS組較對照組顯著增高(P<0.05),Sal A組與對照組比較差異無統計學意義(P>0.05),LPS+Sal A組與對照組比較差異無統計學意義(P>0.05),但低于LPS組(P<0.05)。線粒體膜電位檢測結果顯示:LPS組較對照組顯著降低(P<0.05),Sal A組與對照組比較差異無統計學意義(P>0.05),LPS+Sal A組顯著低于對照組,高于LPS組(P<0.05)。見表1。
2.3 Sal A調節LPS誘導線粒體自噬水平 細胞自噬蛋白檢測結果:Sal A組LC3- II/I、Beclin1、PINK1和Parkin相對表達量與對照組比差異無統計學意義(P>0.05),LPS組LC3- II/I、Beclin1、PINK1和Parkin蛋白相對表達量較對照組均顯著增高(P<0.05),LPS+Sal A組LC3- II/I、Beclin1、PINK1和Parkin蛋白相對表達量與對照組比差異無統計學意義(P>0.05),但低于LPS組(P<0.05)。見表2。線粒體自噬蛋白檢測結果:Sal A組PINK1和Parkin相對表達量與對照組比差異無統計學意義(P>0.05),LPS組PINK1和Parkin蛋白相對表達量與對照組比均顯著增高(P<0.05);LPS+Sal A組PINK1和Parkin蛋白相對表達量與對照組比差異無統計學意義(P>0.05),但低于LPS組(P<0.05)。見表3。
表1 各組ROS含量和線粒體膜電位比較(n=3,±s)
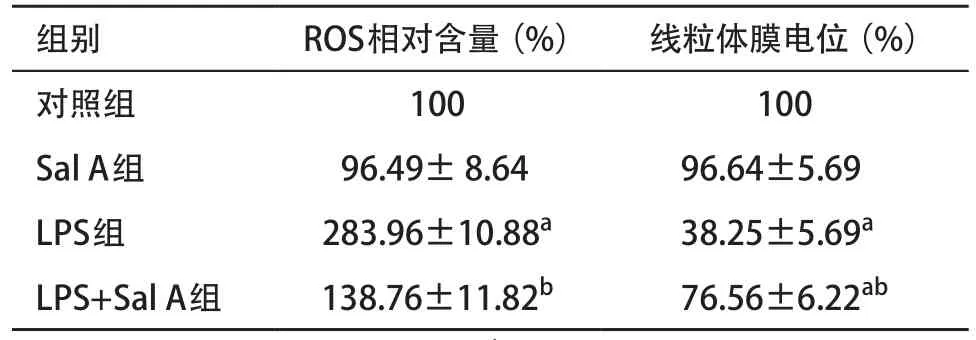
表1 各組ROS含量和線粒體膜電位比較(n=3,±s)
與對照組比:aP<0.05;與LPS組比:bP<0.05
3 討論
LPS誘導的HPMVECs炎性損傷模型是目前常用的體外模擬肺泡-毛細血管屏障損害模型[2]。本研究以該模型作為工具,分析Sal A對LPS誘導細胞炎性損傷的保護作用,結果顯示,10 μg/mL LPS處理HPMVECs 24 h后,細胞相對存活率顯著降低,且隨著時間的延長,細胞相對存活率逐漸下降,說明10 μg/mL LPS成功構建細胞損傷模型;終濃度為0.5、2.5、5、10、25、50 μmol/L的Sal A對HPMVECs無顯著抑制作用。以終濃度為50 μmol/L的Sal A與LPS共同處理HPMVECs后,細胞相對存活率較LPS組顯著增加,說明Sal A具有抑制LPS誘導HPMVECs損傷作用。
線粒體是機體能量代謝的中心,是細胞內ROS的主要來源之一[7]。適量的ROS有利于細胞內信號傳導、基因表達的啟動,促進增殖和分化,但過量ROS誘發氧化應激導致線粒體功能異常、細胞損傷、凋亡[8-9]。有學者認為線粒體損傷初期通過正反饋調節途徑,誘發ROS爆發性增高,導致線粒體膜電位改變;靶向性降低細胞內ROS,可顯著抑制線粒體功能異常[9-10]。本研究中,10 μg/mL LPS處理HPMVECs 48 h后,細胞內ROS含量顯著增高,線粒體膜電位顯著降低,提示LPS誘導線粒體損傷。50 μmol/L的Sal A顯著抑制LPS誘導的細胞內ROS含量增高和線粒體膜電位下降,與文獻[5,11]報道一致。
表2 各組細胞自噬相關蛋白表達比較(n=3,±s)
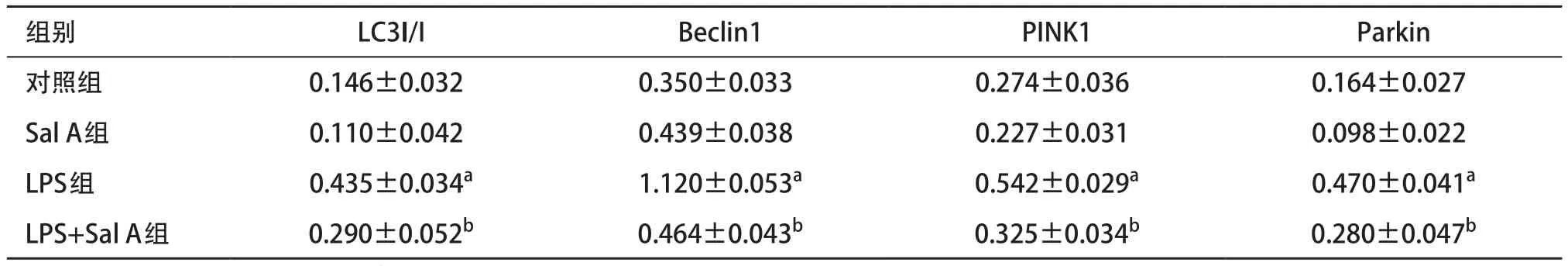
表2 各組細胞自噬相關蛋白表達比較(n=3,±s)
與對照組比:aP<0.05;與LPS組比:bP<0.05
表3 各組線粒體PINK1和Parkin蛋白表達比較(n=3,±s)
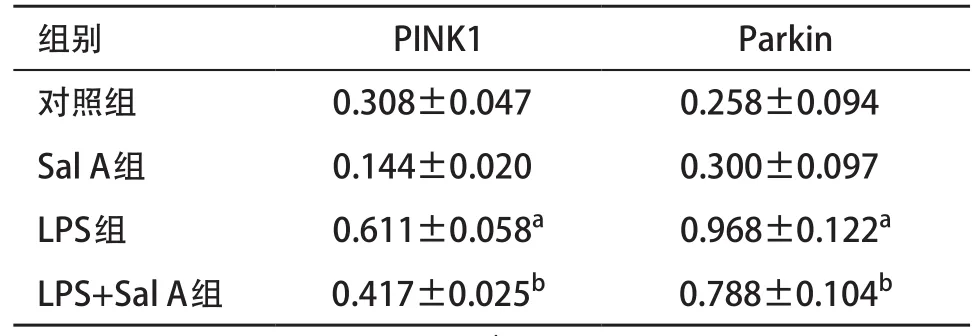
表3 各組線粒體PINK1和Parkin蛋白表達比較(n=3,±s)
與對照組比:aP<0.05;與LPS組比:bP<0.05
線粒體自噬是細胞通過自噬清除損傷線粒體的過程,能夠介導氧化應激引起的細胞損傷[9,12]。ROS增加細胞內PINK1表達,并使PINK1募集Parkin于線粒體上,促進自噬體吞噬損傷的線粒體[13];而氧化清除劑N-乙酰半胱氨酸通過清除細胞內ROS,抑制LC3- II/LC3-I和PINK1表達[14]。FENG等[15]發現抑制過氧硝酸鹽誘導的線粒體自噬能抑制腦缺血再灌注損傷。TANG等[16]發現人參皂苷通過調控線粒體自噬平衡,抑制糖氧缺乏誘導的神經膠質細胞損傷。鄭麗云等[17]研究發現紅景天苷能夠通過線粒體自噬抑制糖氧缺乏星形膠質細胞損傷。本研究結果顯示,與對照組相比,LPS組LC3- II/I、Beclin1、PINK1、Parkin表達顯著增高,說明LPS處理后細胞內自噬水平增加,而用LPS和Sal A共培養后,LC3- II/I、Beclin1、PINK1、Parkin表達降低,說明Sal A拮抗LPS誘導線粒體自噬。進一步提取線粒體蛋白,證實Sal A顯著降低LPS誘導的PINK1、Parkin表達。
綜上所述,本研究證實Sal A對LPS誘導的HPMVECs具有保護作用,但由于實驗過程中未應用陽性對照,僅反映了Sal A降低了LPS誘導的ROS含量,穩定了線粒體膜電位和降低了線粒體自噬水平,仍無法證實三者及其相互關系在Sal A抑制LPS誘導HPMVECs損傷中的作用機制,仍需進一步研究。
參考文獻:
[1] BERNARD G. Acute lung failure-our evolving understanding of ARDS[J]. N Engl J Med, 2017, 377(6): 507-509.
[2] ZOU Y, BAO S, WANG F, et al. FN14 blockade on pulmonary microvascular endothelial cells improves the outcome of sepsis-induced acute lung injury[J]. Shock, 2018, 49(2):213-220.
[3] STANDIFORD T J, WARD P A. Therapeutic targeting of acute lung injury and acute respiratory distress syndrome[J].Transl Res, 2016, 167(1): 183-191.
[4] FAN H Y, YANG M Y, QI D, et al. Salvianolic acid A as a multifunctional agent ameliorates doxorubicin-induced nephropathy in rats[J]. Sci Rep, 2015, 5: 12273.
[5] CHEN Y C, YUAN T Y, ZHANG H F, et al. Salvianolic acid A attenuates vascular remodeling in a pulmonary arterial hypertension rat model[J]. Acta Pharmacol Sin, 2016, 37(6):772-782.
[6] 邱偉文, 鄭麗云, 鄔至平, 等. 腦心通對氧化低密度脂蛋白誘導血管平滑肌細胞增殖和自噬的影響[J]. 溫州醫科大學學報, 2016, 46(5): 340-343.
[7] SHADEL G S, HORVATH T L. Mitochondrial ROS signaling in organismal homeostasis[J]. Cell, 2015, 163(3): 560-569.
[8] ROWLANDS D J. Mitochondria dysfunction: A novel therapeutic target in pathological lung remodeling or bystander?[J]. Pharmacol Ther, 2016, 166: 96-105.
[9] LEE J, GIORDANO S, ZHANG J. Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling[J].Biochem J, 2012, 441(2): 523-540.
[10] GUIDARELLI A, FIORANI M, CERIONI L, et al. Arsenite induces DNA damage via mitochondrial ROS and induction of mitochondrial permeability transition[J]. Biofactors,2017, 43(5): 673-684.
[11] MAO K, SHU W, QIU Q, et al. Salvianolic acid A protects retinal pigment epithelium from OX-LDL-induced inflammation in an age-related macular degeneration model[J].Discov Med, 2017, 23(125): 129-147.
[12] PIANTADOSI C A, SULIMAN H B. Mitochondrial dysfunction in lung pathogenesis[J]. Annu Rev Physiol, 2017,79: 495-515.
[13] XIAO B, GOH J Y, XIAO L, et al. Reactive oxygen species trigger Parkin/PINK1 pathway-dependent mitophagy by inducing mitochondrial recruitment of Parkin[J]. J Biol Chem,2017, 292(40): 16697.
[14] WEI X, QI Y, ZHANG X, et al. Cadmium induces mitophagy through ROS-mediated PINK1/Parkin pathway[J]. Toxicology Methods, 2014, 24(7): 504-511.
[15] FENG J, CHEN X, GUAN B, et al. Inhibition of peroxynitrite-induced mitophagy activation attenuates cerebral is chemia-reperfusion injury[J]. Mol Neurobiol (2018). https://doi.org/10.1007/s12035-017-0859-x.
[16] TANG X, CHEN N, LONG X. Ginsenoside Rg1 improves is chemic brain injury by balancing mitochondrial biogenesis and mitophagy[J]. Trop J Pharm Res, 2017, 16(10): 2469-2475.
[17] 鄭麗云, 黃慧芬, 邱偉文. 紅景天苷對缺氧缺糖誘導的星形膠質細胞線粒體自噬的影響[J]. 溫州醫科大學學報,2017, 47(10): 744-747.
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
