Orbitrap高分辨質譜法高通量篩查生化藥品、中成藥與保健品中非法添加的13種消化類化學藥物
2018-04-18 03:22:13郭常川張迅杰賀美蓮
分析測試學報 2018年3期
關鍵詞:藥品
郭常川,孫 華,石 峰*,邢 晟,張迅杰,李 可,賀美蓮,姜 瑋,3*
(1.山東省食品藥品檢驗研究院,山東 濟南 250101;2.山東大學 藥學院,山東 濟南 250012;3.山東大學 化學與化工學院,山東 濟南 250100)
消化不良是一種臨床癥候群,主要由胃動力障礙所引起,包括胃蠕動差的胃輕癱和食道反流病,是一種發病人群很廣的常見疾病。近年來,市面上出現很多針對消化不良癥狀的聲稱“純天然”、“無化學添加”的生化藥品、中成藥或保健品。然而,此類產品質量良莠不齊,一些不法分子為片面追求治療效果、獲取經濟暴利,擅自在其中添加化學藥品,不僅違反了《藥品管理法》和《食品安全法》的規定,還嚴重危及廣大人民群眾的身體健康。中成藥、生化藥品或保健品若在說明書中明確注明含有某種化學成分,則符合法律法規;若未注明卻被檢出,則為非法添加。因此,為嚴厲打擊非法添加行為,保證人民群眾的食品和用藥安全,亟需開發快速、準確、高靈敏、高選擇性的用于非法添加消化類化學藥物的快速篩查方法。
當前,已報道的非法添加檢測手段以液質聯用法(LC-MS)[1-3]為主,也有關于高效液相色譜法(HPLC)[4]、薄層色譜法(TLC)-表面增強拉曼(SERS)聯用法[5-6]、膠束電動色譜法[7]的報道。Orbitrap高分辨質譜儀(Orbitrap HRMS)是最新的高分辨質譜技術,分辨率可達1 000 000以上,質量精度可達0.1~1 ppm[8],可進行高精度的目標物或非目標物的篩查。近年來,Orbitrap 高分辨質譜技術的應用日益增多,已用于食品安全[9-10]、藥品安全[11]、中成藥成分分析[12]、藥物動力學[13]、蛋白組學[14]、代謝組學[15]研究等領域。
本文首次將超高效液相色譜-Orbitrap高分辨質譜聯用(UHPLC-HRMS)技術用于消化類化學藥物非法添加的快速篩查、鑒定和定量研究。通過Full MS/dd-MS2(Data-dependent MS2) 模式,在一個分析周期(10 min)內完成對樣品的高精度一級、二級掃描,得到準確質量數和準確碎片離子信息,同時對潛在的陽性樣品進行定量分析。本方法具有快速、準確、高靈敏度、高選擇性的特點,為打擊非法添加行為提供了新手段、新技術。
1 實驗部分
1.1 儀器與試劑
Thermo Q Exactive PlusTM超高效液相色譜-質譜聯用系統包括Ultimate 3000液相泵、自動進樣器、柱溫箱以及Orbitrap高分辨質譜部分(Thermofisher Scientific,德國);XCalibur 4.0軟件(Thermofisher Scientific,美國)用以質譜儀控制和數據處理;Mettler XS205電子天平(Mettler Toledo,瑞士);KQ-300 GDV溫控超聲儀(昆山舒美,中國);Sigma 3K15高速冷凍離心機(Sigma-Aldrich,德國);IKA Vortex3旋渦混勻器(IKA,德國)。色譜柱為Thermo Hypersil Gold C18(2.1 mm×100 mm,3 μm,Thermofisher Scientific,美國)。
對照品左卡尼汀、法莫替丁、西咪替丁、鹽酸雷尼替丁、消旋山莨菪堿、甲氧氯普胺、奧美拉唑、雷貝拉唑鈉、泮托拉唑鈉、多潘立酮、西沙比利和枸櫞酸莫沙必利均購自中檢院,伊托必利購自加拿大TRC公司;HPLC級甲醇、乙腈以及超純水均購自Fisher公司(美國),HPLC級甲酸、甲酸銨、乙酸銨購自ACS恩科化學公司(美國);樣品來源:156批生化藥品(復方胃蛋白酶顆粒)來自2017年國家評價性抽驗檢品,38批中成藥、43批保健品來自山東省食品藥品監督管理局或山東省各市、縣食品藥品監督管理局抽樣。將未檢出以上13種成分的10批樣品混合均勻,作為陰性樣品基質。10批陰性樣品編號分別為YC201700004,YC201700025,YC201700039,YC201700040,YC201700055,YC201700057,YC201700058,YC201700068,YC201700114,YC201700117。
1.2 溶液的制備
1.2.1對照品溶液的制備精密稱取各對照品適量,置于25 mL容量瓶中,加甲醇溶解并定容至刻度,搖勻,作為對照品儲備液(質量濃度為200 mg/L)。向空白基質中加標準品,配成相當于實際樣品質量濃度為8、40、200、400、800 μg/L的標準溶液和QC(質量控制)溶液。
1.2.2供試品溶液的制備取供試品約0.5 g,精密稱定,置于10 mL容量瓶中,加提取溶劑甲醇-水(50∶50,體積比)適量,超聲提取15 min,冷卻至室溫,加提取溶劑定容至刻度,上清液過濾膜,取續濾液即得供試品溶液。
1.3 液相色譜-質譜聯用條件
1.3.1色譜條件色譜柱為Thermo Hypersil Gold C18(2.1 mm ×100 mm,3 μm)。流動相:0.1%甲酸水溶液(A)-乙腈(B),梯度洗脫程序:0~1.0 min,5%B;1.0~5.0 min,5%~85%B;5.0~7.5 min,85%B;7.5~7.6 min,85%~5%B;7.6~10.0 min,5%B。流速:300 μL·min-1;柱溫:40 ℃;自動進樣器溫度:20 ℃;進樣量:5 μL。
1.3.2質譜條件Q ExactiveTM質譜系統配有HESI離子源,采用正離子模式,噴霧電壓為3.0 kV,毛細管和噴霧溫度分別為350 ℃和250 ℃。鞘氣和輔助氣壓力分別設為40、15 arb,S-lens RF電壓為50 V。噴霧氣和碰撞氣均為氮氣。使用校正溶液(含咖啡因、四肽MRFA和Ultramark 1621的混合溶液)每3 d校正1次質量軸。掃描方式采用正離子Full MS/dd-MS2模式。Full MS一級全掃描范圍為m/z100~1 000,自動增益控制AGC、最大注入時間IT分別設為1.0e6和100 ms;dd-MS2數據依賴的二級掃描AGC設為2.0e5,最大IT設為50 ms,分離窗口設為1.0m/z。各化合物的歸一化碰撞能量設為20%、40%、60%。
2 結果與討論
2.1 方法的建立
考慮到13種化合物的極性差異較大,故選擇甲醇-水(體積比50∶50)作為提取溶劑,結果表明其對13種化合物均有較高的提取效率,且濾液潔凈,無需進一步凈化即可直接進樣,簡化了樣品前處理。
由于13種化合物結構中均含有較多的N原子,正離子質譜掃描模式下響應高,因此選擇正離子模式。分別將13種化合物標準溶液流動注入質譜儀進行參數優化,獲得相應的準分子離子峰,輸入至方法設置中。各化合物二級質譜掃描的碰撞能量統一設為20%、40%、60%,得到的二級質譜圖是在3種碰撞能量下二級質譜圖的疊加,有效避免了采用單一碰撞能量可能導致的不同儀器間重現性差的情況。在建立的LC-MS聯用方法中,掃描方式選擇正離子Full MS/dd-MS2模式,此模式包含一次一級全掃描和一次數據依賴的二級掃描。一級質譜全掃描采用70 000 FWHM分辨率,確保得到高分辨質譜準確質量數;二級質譜掃描采用17 500 FWHM,保證高分辨掃描的同時加快掃描速度。
對比了甲醇-水和乙腈-水混合體系作為流動相的效果,并在水相中分別添加了少量甲酸、甲酸銨、乙酸銨等。結果表明,乙腈-0.1%甲酸水體系能得到最佳的分離效果,獲得最佳的色譜峰形和較高的靈敏度,因此選擇乙腈-0.1%甲酸水體系作為流動相,并采用“1.3.1”所述梯度洗脫程序。
2.2 專屬性
稱取陰性樣品適量,按“1.2.2”方法處理得到空白基質溶液,進樣5 μL分析,空白基質溶液的提取離子色譜圖顯示,各化合物保留時間處未出現干擾峰或干擾峰很小(遠低于檢出限),對定量分析沒有影響,專屬性強。單次測定在10 min內即可完成,分析效率高,尤其適用于高通量篩查。另取200 μg/L的QC溶液進樣5 μL分析,記錄色譜圖(圖1)。
化合物的保留時間、準確質量數和碎片離子信息如表1所示。理論質量數與實際測得的質量數相對偏差越小,表明篩查結果的可信程度越高。表1中13種分析物的實測和理論質量數偏差均不超過3.08 ppm,顯示了Orbitrap HRMS高度可靠的質量精度。代表性化合物雷尼替丁的二級質譜圖及裂解機理如圖2所示,二級質譜碎片離子輔助確證化合物的存在,裂解機理闡明了主要碎片離子的化學結構及裂解途徑。
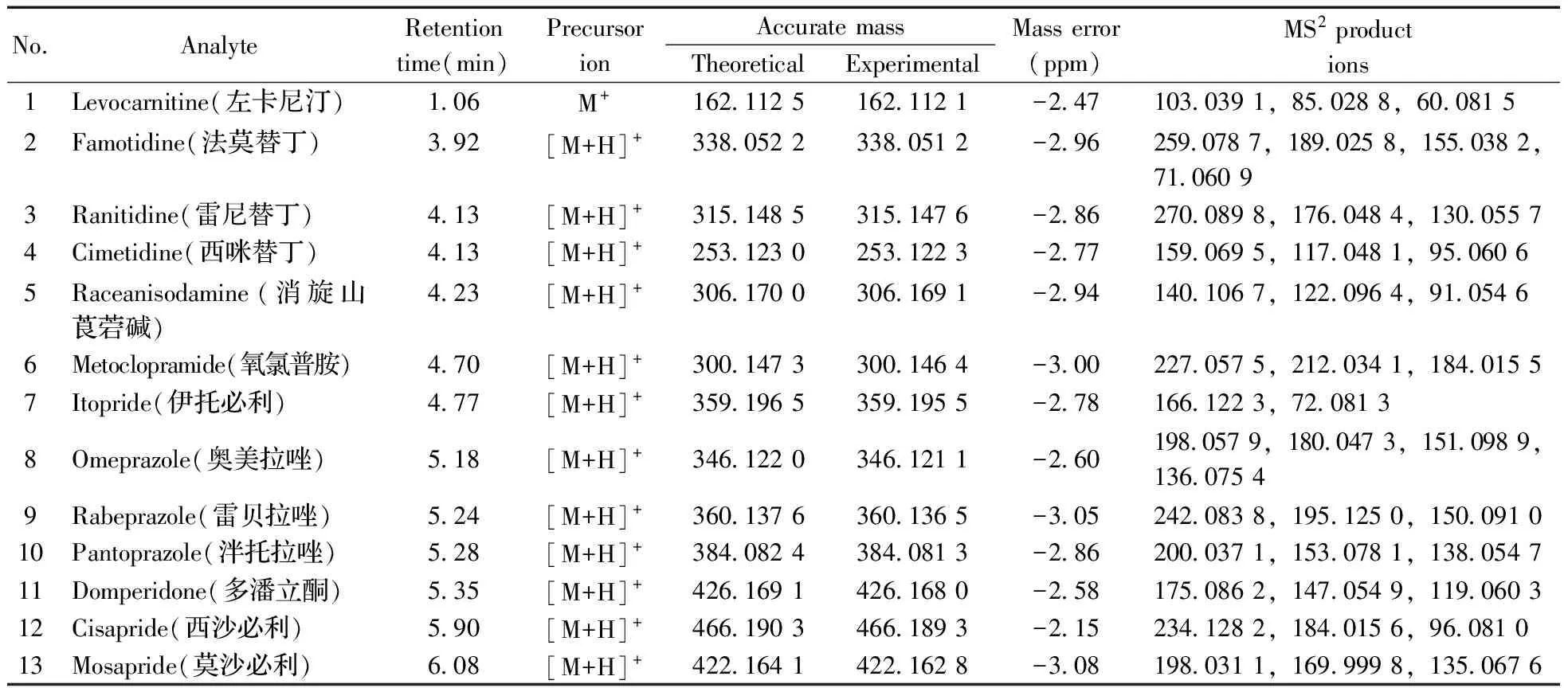
表1 消化類藥物的保留時間和準確質量數測定Table 1 Retention time and accurate mass of digestive system drugs
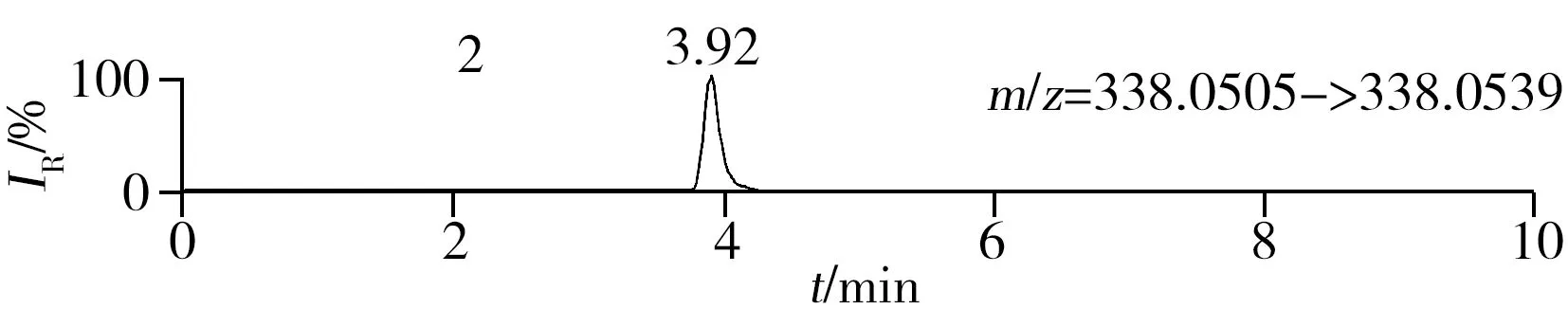
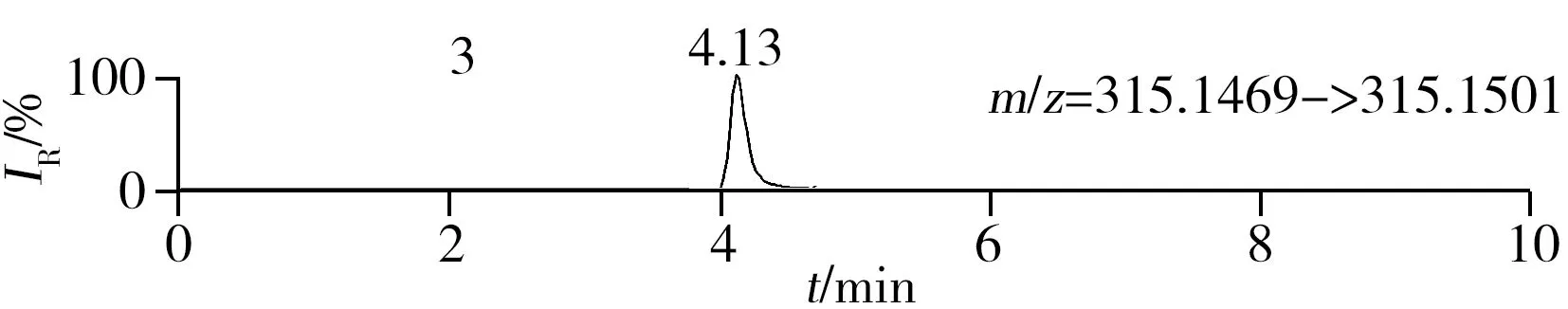
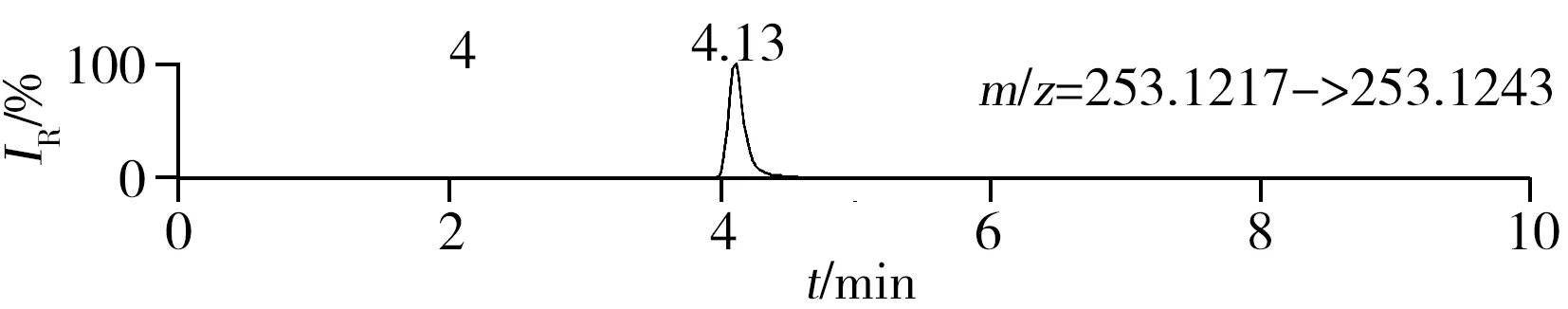
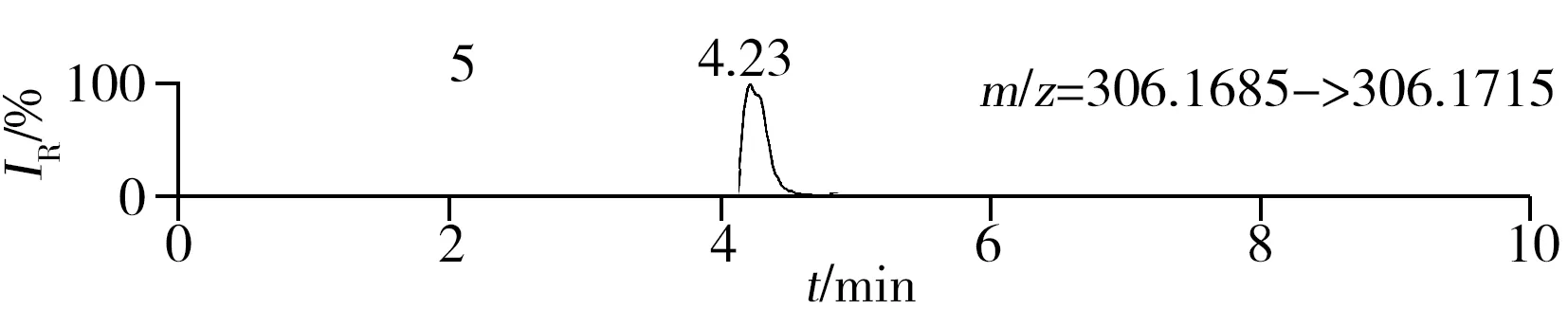
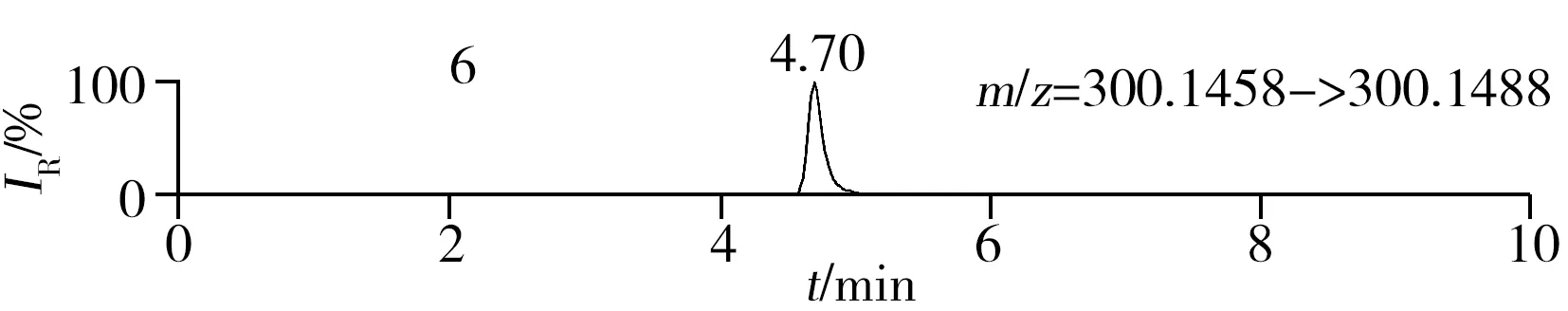
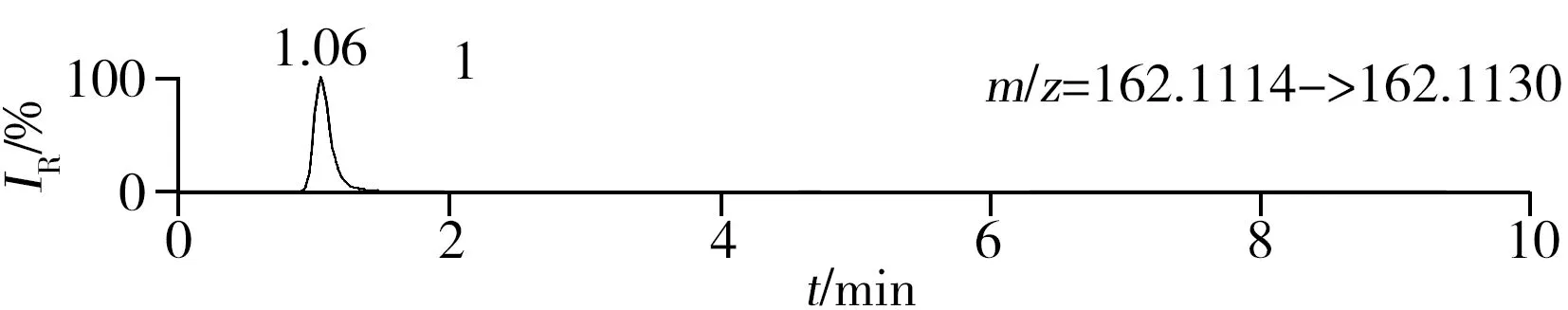
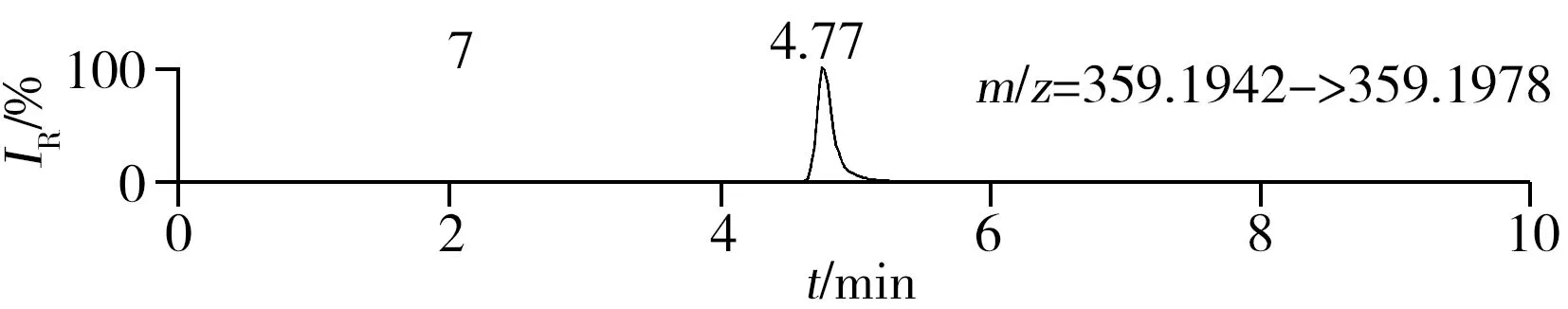
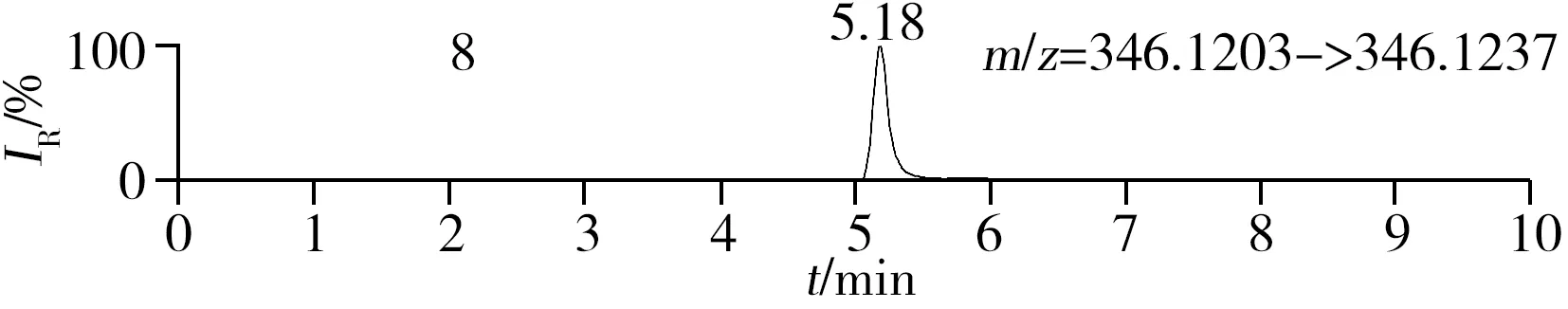
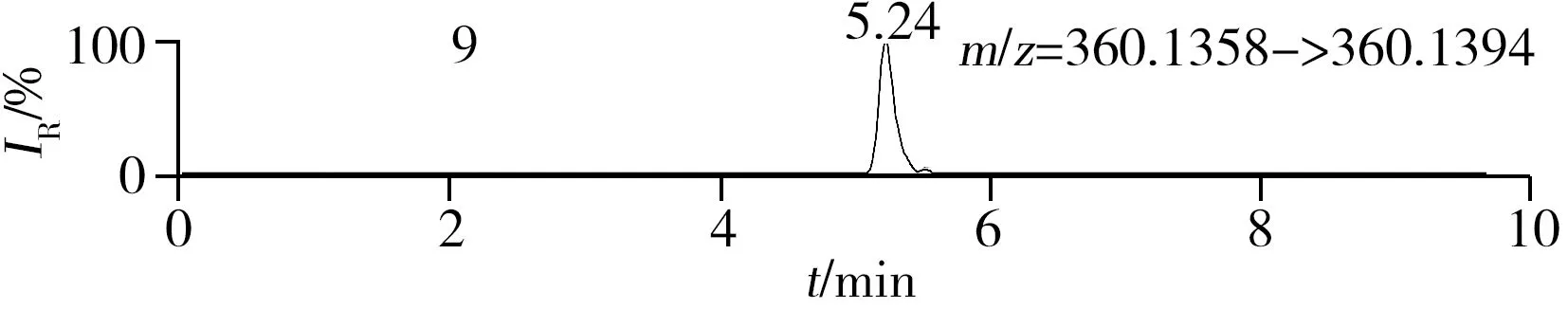
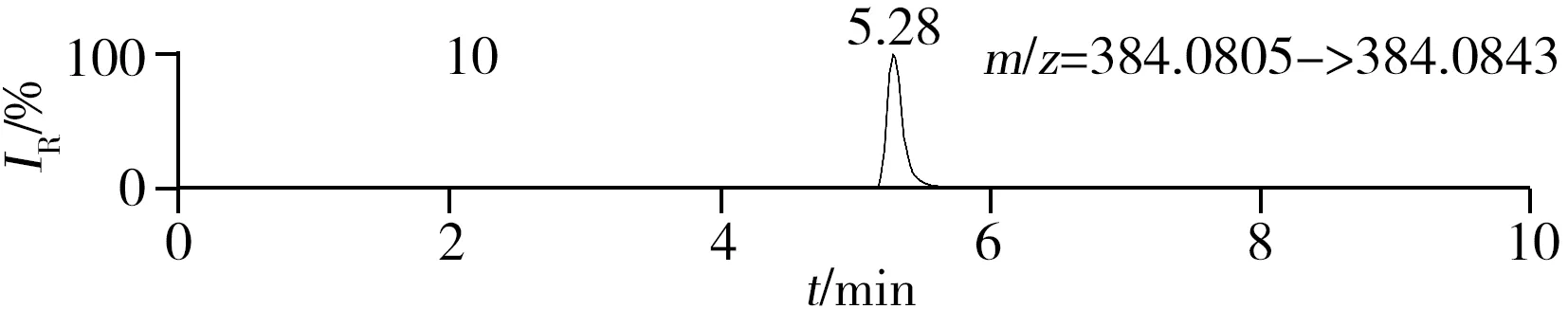
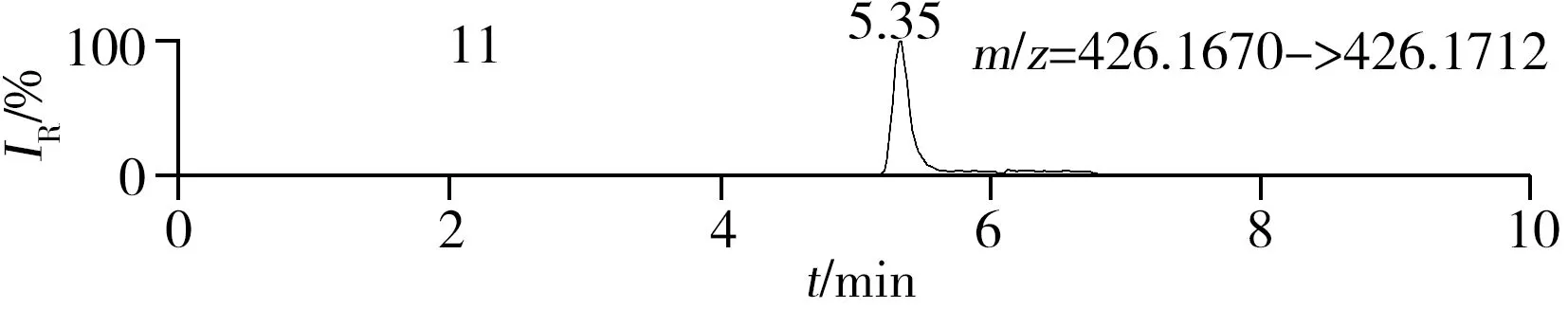
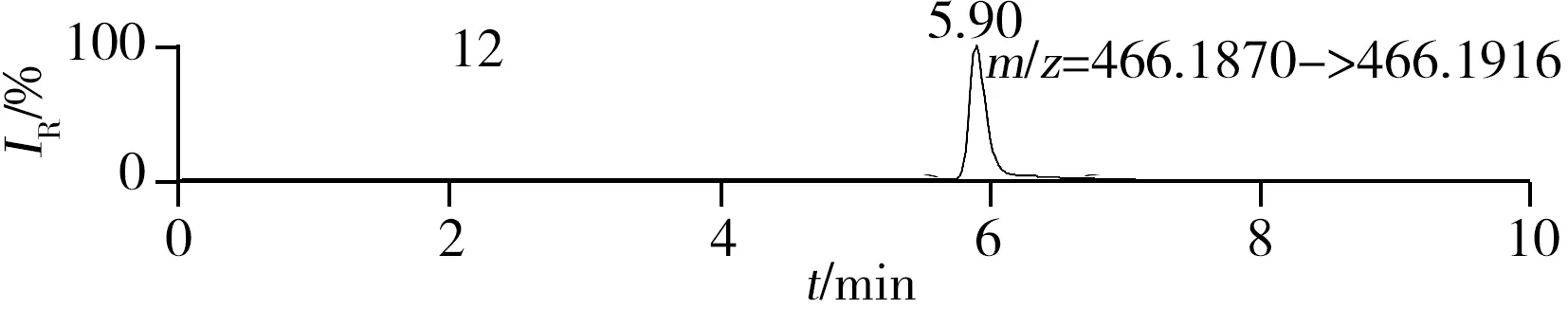
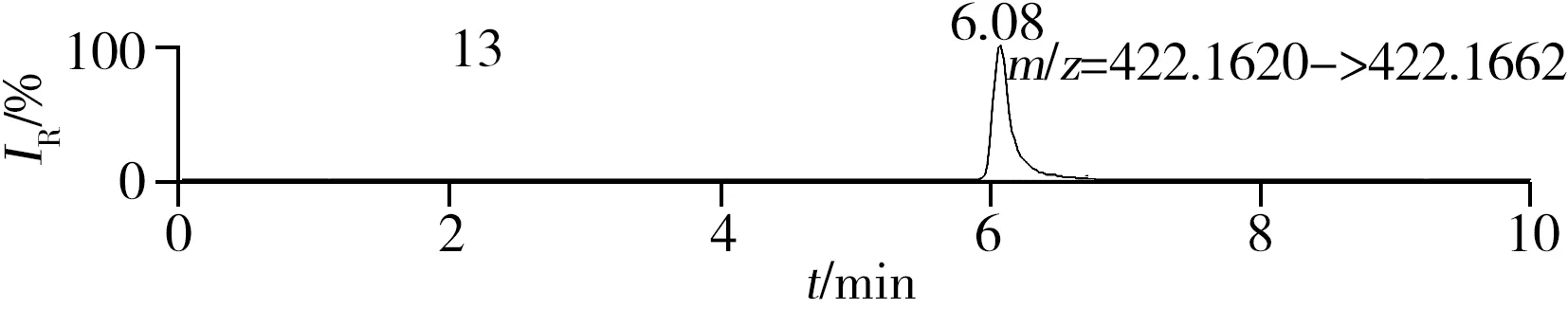
圖1 QC溶液(200 μg/L)的提取離子色譜圖Fig.1 Extracted ion chromatograms of digestive drugs in 200 μg/L spike solutionthe noted numbers were the same as those in Table 1
2.3 檢出限
逐級稀釋QC溶液并進樣分析,記錄色譜圖,按照3倍信噪比計算檢出限。結果表明各化合物的檢出限均為1.0 μg/L。
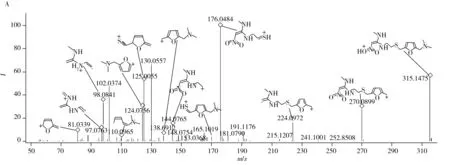
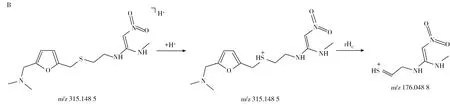
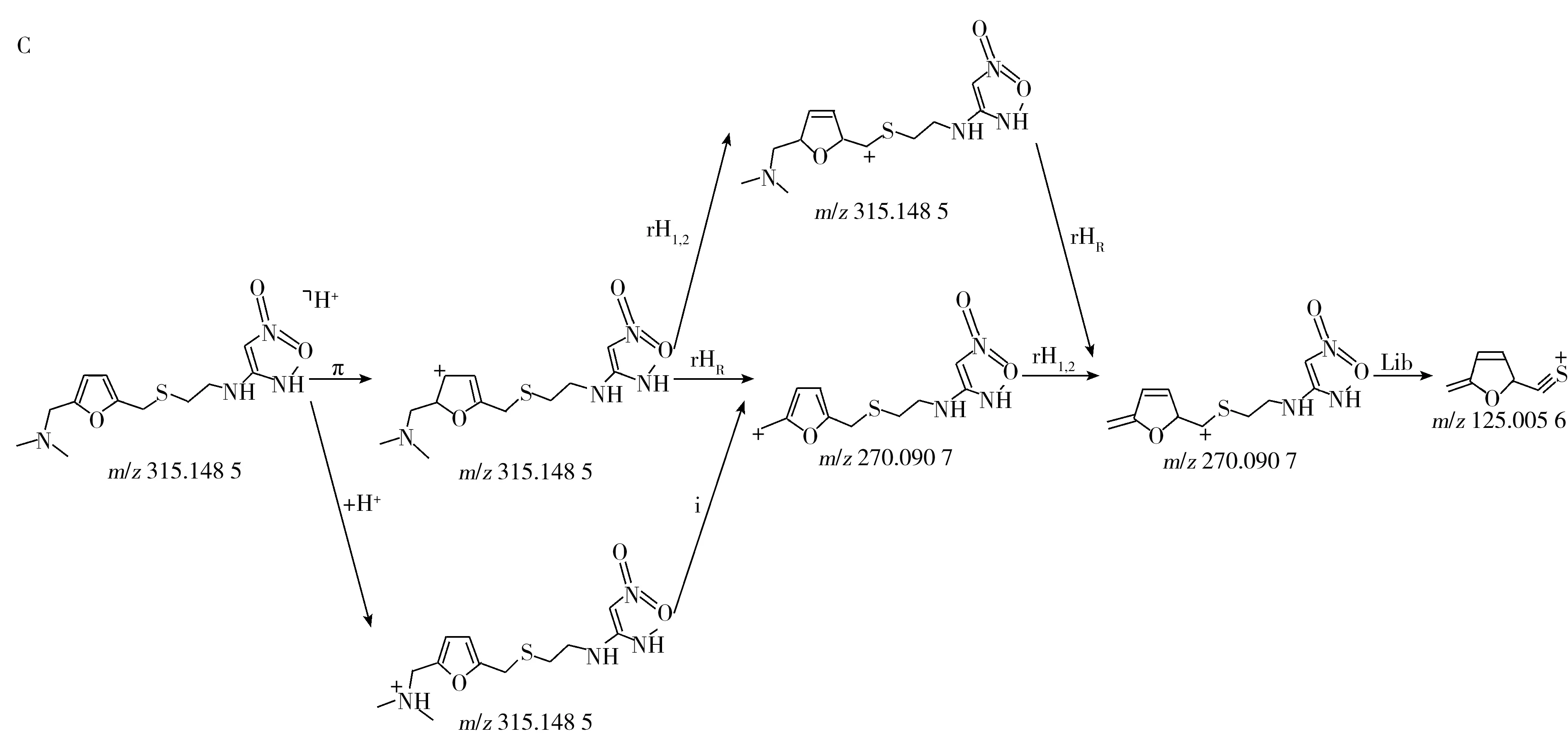
圖2 雷尼替丁的二級質譜圖(A)以及碎片離子m/z 176.048 8(B)與m/z 270.090 7及m/z 125.005 6(C)的裂解機理Fig.2 MS2 mass spectra(A) and fragmention mechanisms of fragment ions m/z 176.048 8(B),m/z 270.090 7 and m/z 125.005 6(C)
2.4 線性范圍與定量下限
按“1.2.1”配制系列標準溶液,進樣5 μL測定峰面積。以峰面積(y)對標準溶液質量濃度(x,μg/L)擬合校正曲線,建立回歸方程。結果表明,13種化合物的峰面積與質量濃度線性關系良好,線性范圍均為8.0~800 μg/L,相關系數(r)為0.998 5~1.000 0(表2)。以線性范圍最低點作為定量下限,13種化合物的定量下限均為8.0 μg/L。
2.5 回收率、準確度與精密度
分別精密移取對照品儲備液10 μL、陰性供試品0.5 g,置于10 mL容量瓶中,加提取溶劑適量,超聲提取15 min,冷卻至室溫,加提取溶劑定容至刻度,上清液過濾膜,取續濾液即得供試品溶液(質量濃度約為200 μg/L),平行制備6份。進樣,按擬定方法測定回收率,計算平均回收率及相對標準偏差(RSD)。回收率平均值用于評價準確度,RSD值用于評價精密度,實驗結果見表2,13種化合物的回收率為94.6%~106.2%,RSD為1.2%~2.9%。
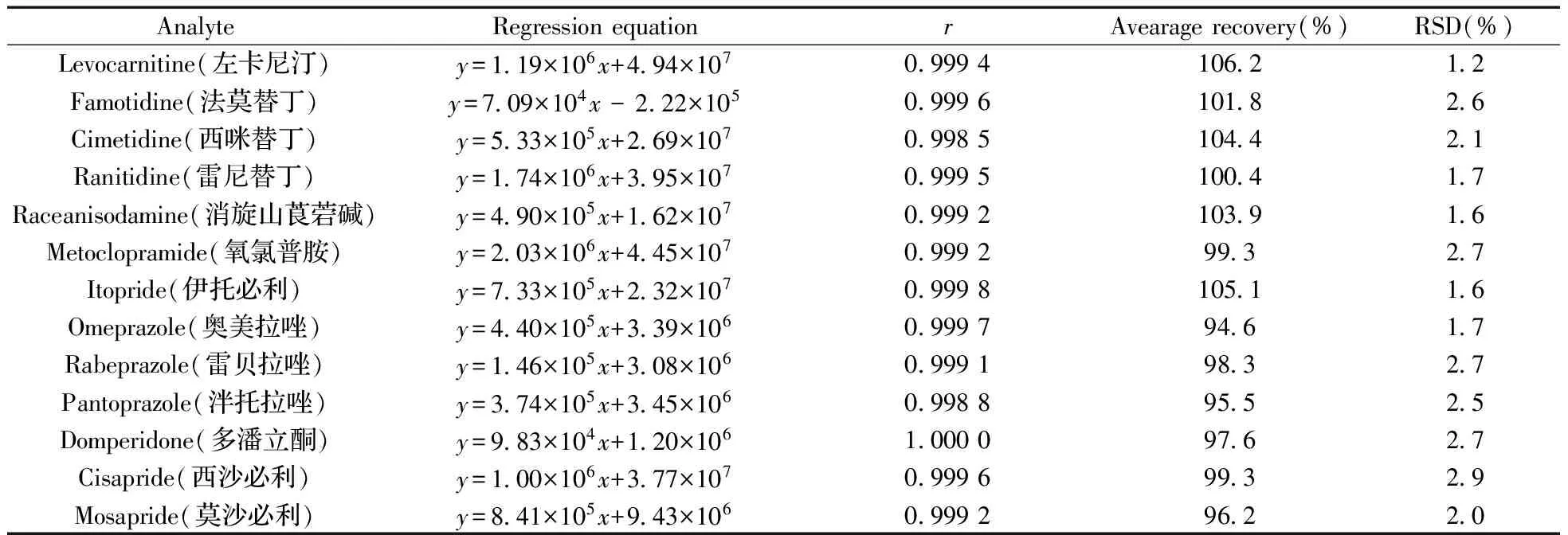
表2 13種消化類化學藥物的回歸方程,相關系數和回收率Table 2 Regression equation,correlation coefficient(r) and recovery of digestive system drug
2.6 基質效應與穩定性
取200 μg/L的QC溶液和200 μg/L的純溶劑對照品溶液,分別進樣,計算響應值百分比,即為基質效應。結果表明13種化合物的基質效應均在92.3%~105.6%之間,表明空白基質對化合物響應影響很小。
用空白基質配制200 μg/L的標準溶液,分別在1、2、4、6、8、12、24 h進樣,以各化合物的提取離子峰面積為指標,代入標準曲線計算濃度與實際濃度的比值(穩定性)在94.5%~102.8%之間,由此可見配制的標準溶液在24 h內穩定。
2.7 實際樣品分析
將建立的高分辨質譜法用于156批生化藥品、38批中成藥、43批保健品中非法添加消化類化學藥物的篩查。比較供試品和對照品的提取離子色譜圖、一級質譜和二級質譜,若供試品某成分的保留時間、一級質譜準確質量、碎片離子準確質量均與對照品一致,則判定該成分為非法添加物。
經篩查發現,156批生化藥品均未檢出左卡尼汀等13種化學藥物,而5批中成藥中檢出西咪替丁(含量為1.4~17.9 mg/g),3批保健品中檢出多潘立酮(含量8.61~14.3 mg/g),代表性陽性樣品(檢出多潘立酮)圖譜見圖3所示。以上結果表明消化類化學藥物非法添加在中成藥和保健品中較為突出。另一方面,156批生化藥品均未檢出非法成分,可能與其來源是正規藥品企業有關。以上結果表明本文所建立的高通量HRMS法具有良好的可靠性。
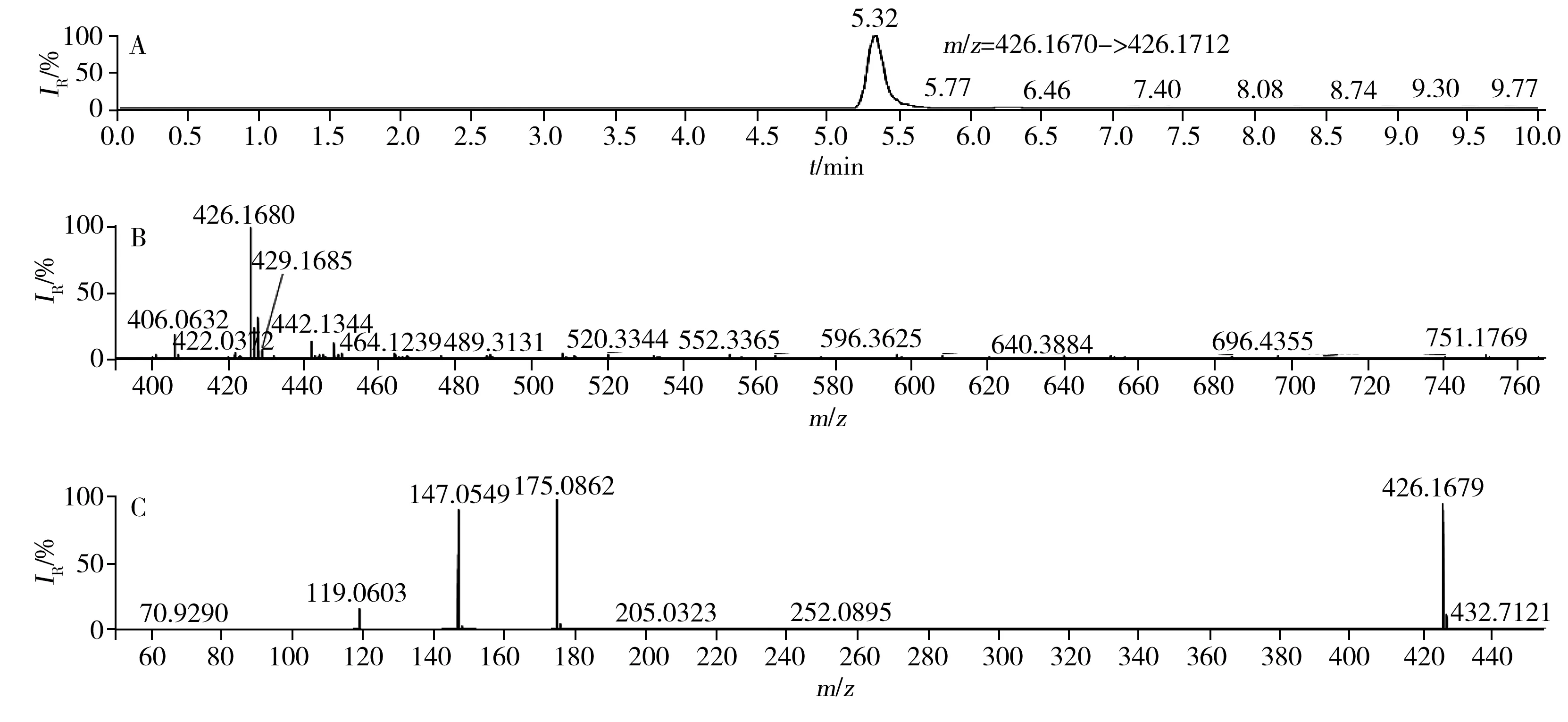
圖3 代表性陽性樣品YW201708461的提取離子色譜圖(A)、一級質譜圖(B)和二級質譜圖(C)Fig.3 Extracted ion chromatogram(A),MS(B) and MS2(C) spectra of digestive drugs in representative positive sample YW201708461
3 結 論
本研究建立了一種快速篩查非法添加消化類化學藥物的方法,可在10 min 內同時測定13種消化類藥物。該法選擇性好、靈敏度高、快速簡便、可操作性強、準確可靠,極大地提高了檢測效率,適用于生化藥品、中成藥及保健品非法添加檢測,已用于日常檢測工作中,為監管相關非法添加提供了有力的技術支持。
參考文獻:
[1] Choi J Y,Heo S,Yoo G J,Park S K,Yoon C Y,Baek S Y.FoodAddit.Contam.:A,2015,32(7):1029-1039.
[2] Heo S,Choi J Y,Yoo G J,Park S K,Baek S Y.Biomed.Chromatogr.,2017,31(4):6.
[3] Kim E H,Seo H S,Ki N Y,Park N H,Lee W,Do J A,Park S,Baek S Y,Moon B,Oh H B,Hong J.J.Chromatogr.A,2017,1491:43-56.
[4] Huang Y C,Lee H C,Lin Y L,Li C Y,Tsai C F,Cheng H F.FoodAddit.Contam.:A,2016,33(2):179-185.
[5] Li H,Zhu Q X,Chwee T S,Wu L,Chai Y F,Lu F,Yuan Y F.Anal.Chim.Acta,2015,883:22-31.
[6] Lv D Y,Cao Y,Lou Z Y,Li S J,Chen X F,Chai Y F,Lu F.Anal.Bioanal.Chem.,2015,407(5):1313-1325.
[7] Maier V,Znaleziona J,Jirovsky D,Skopalova J,Petr J,Sevcik J.J.Chromatogr.A,2009,1216(20):4492-4498.
[8] Zubarev R A,Makarov A.Anal.Chem.,2013,85(11):5288-5296.
[9] Jia W,Shi L,Chu X.FoodChem.,2018,239:427-433.
[10] Marín-Sáez J,Romero-González R,Garrido F A.J.Chromatogr.A,2017,1518:46-58.
[11] Cheng Q,Shou L,Chen C,Shi S,Zhou M.J.Chromatogr.B,2017,1064:92-99.
[12] Wang S S,Xu H Y,Ma Y,Wang X G,Shi Y,Huang B,Tang S H,Zhang Y,Li D F,Liang R X,Yang H J.J.Pharm.Biomed.Anal.,2015,111(Suppl C):104-118.
[13] Zuo L,Zhong Q,Wang Z,Sun Z,Zhou L,Li Z,Xu T,Shi Y,Tang J,Du S,Zhang X.J.Pharm.Biomed.Anal.,2017,146:347-353.
[14] Li X,Shi F,Gong L,Hang B,Li D,Chi L.Int.J.Nanomed.,2017,12:4443-4454.
[15] Huang X H,Guo C N,Chen Z L,Liu Y H,He L M,Zeng Z L,Yan C Q,Pan G F,Li S P.J.Chromatogr.B,2015,1000:147-154.
猜你喜歡
中國合理用藥探索(2022年1期)2022-11-26 00:22:32
世界最新醫學信息文摘(2021年12期)2021-06-09 08:36:56
小學生優秀作文(低年級)(2018年6期)2018-05-19 01:54:28
消費導刊(2017年20期)2018-01-03 06:27:16
中國衛生(2016年6期)2016-11-23 01:09:08
中國衛生(2016年5期)2016-11-12 13:25:28
中國藥物應用與監測(2015年5期)2015-12-11 03:15:54
中國衛生(2015年9期)2015-11-10 03:11:14
中國衛生(2015年5期)2015-11-08 12:09:48
中國衛生(2015年4期)2015-11-08 11:15:58
