氯化鉀溶液對大鼠腦組織缺血再灌注損傷的影響及機制
2018-04-11 05:02:51方衛李諾覃斯娜覃濤楊葉桂陳蒙華
山東醫藥 2018年10期
關鍵詞:模型
方衛,李諾,覃斯娜,覃濤,楊葉桂,陳蒙華
(廣西醫科大學附屬第二醫院,南寧530000)
腦缺血持續一段時間后恢復血供,腦細胞可能因為缺血再灌注而遭受更嚴重的損傷,其機制與細胞凋亡的誘發有關[1]。有研究表明,細胞凋亡與細胞內K+濃度具有密切關系。體外實驗發現,在細胞凋亡初期即可觀察到K+外流,而提高細胞培養液的K+濃度能夠減輕、逆轉細胞凋亡[2]。Ca2+作為細胞的第二信使參與腦內的多種代謝活動,發生缺血再灌注損傷時,細胞內的Ca2+急劇上升,超過Ca2+的調控范圍,造成Ca2+超載以及一系列的級聯反應;也有研究[3]表明,使用鈣通道阻滯劑可以減輕細胞凋亡。因此我們推測,提高細胞外K+濃度可能部分通過拮抗Ca2+超載產生了保護作用。為驗證此假說,2017年3~7月,我們借助大腦中動脈栓塞(MCAO)模型,在大鼠腦組織恢復灌注時給予氯化鉀(KCl)溶液調整血K+濃度,觀察局部腦組織損傷情況,并對Ca2+超載相關指標進行檢測。
1 材料與方法
1.1 動物與試劑
SPF級健康雄性SD大鼠36只,體質量220~280 g,由廣西醫科大學實驗動物中心提供。大鼠術前禁食12 h,自由飲水。鈣ATP酶(Ca2+-ATPase)、乳酸脫氫酶(LDH)、鈣調神經磷酸酶(CaN)、Ca2+檢測試劑盒均購自南京建成生物工程研究所,鈣調蛋白(CAM)檢測試劑盒購自武漢華美生物工程有限公司。
1.2 動物分組
將大鼠隨機分成模型1組、模型2組和假手術組各12只。
1.3 模型制備與干預
模型1組和模型2組腹腔注射10%水合氯醛麻醉,從大鼠胸鎖乳突肌和頸前肌群之間向深部分離,顯露右側頸總動脈、頸內動脈和頸外動脈。使用動脈夾夾住頸總動脈近心端,縫線結扎頸外動脈遠心端。在頸外動脈距離頸總動脈分叉約5 mm處剪口,用線栓由剪口處經頸外動脈進入頸內動脈,剪斷頸外動脈并游離,調整方向避開翼腭動脈的開口處到達大腦中動脈。當線栓進入長度距頸總動脈分叉處15 mm時,緩慢插入線栓,使線栓頭部剛好完全阻閉右側大腦中動脈入口。阻閉120 min后,拔出線栓并移除頸總動脈的血管夾,使缺血部位恢復灌注;同時模型1組由右側頸外靜脈泵入2.8%KCl溶液90 μg/g,模型2組由右側頸外靜脈泵入生理鹽水90 μg/g。假手術組只分離和結扎右側頸外動脈,不使用線栓阻斷大腦中動脈。恢復灌注后監測大鼠生命體征1 h,隨后縫合傷口,將大鼠放回籠內單獨喂養。
1.4 神經功能評價
再灌注24 h后,各組隨機抽取6只大鼠,依照文獻[4]對大鼠進行神經功能評分。評分標準:0分為正常,無神經損傷癥狀;1分為不能完全伸展對側前爪;2分為向外側轉圈;3分為向對側傾倒;4分為不能自發行走,意識喪失。
1.5 腦組織病理檢查
采用HE染色法。再灌注24 h后,各組隨機取6只大鼠。用10%水合氯醛麻醉,斷頭取腦;4%多聚甲醛灌注固定,石蠟包埋,常規脫蠟水化,梯度乙醇浸洗。蘇木精染色,自來水沖洗;鹽酸乙醇分化,自來水沖洗;伊紅染色,自來水沖洗;逆乙醇梯度脫水,二甲苯再次透明,封片。光學顯微鏡下觀察腦組織病理學變化,并拍照記錄。
1.6 腦組織上清液Ca2+超載相關指標檢測
采用比色法。再灌注24 h后,各組取剩余6只大鼠,麻醉,斷頭,取右側腦皮質部分制成勻漿,離心取上清液。Ca2+-ATPase、LDH、CaN活性檢測以及Ca2+水平測定采用化學比色法,CAM檢測采用ELISA法。
1.7 統計學方法
2 結果
2.1 三組神經功能評分比較
模型1組、模型2組、假手術組的神經功能評分分別為(2.17±0.75)、(3.50±0.55)、(0.83±0.41)分。與假手術組比較,模型2組的神經功能評分高(P<0.05);與模型2組比較,模型1組的神經功能評分低(P<0.05)。
2.2 三組腦組織病理結果比較
與假手術組比較,模型2組低倍鏡下可見明顯的低密度區,且集中于右側皮質部分,此低密度區域正常細胞數量極少;高倍鏡下可見多數細胞發生變性,壞死,可見染色質的邊集、核固縮,空泡細胞也較多見。與模型2組比較,模型1組低倍鏡下可見右側皮質部分低密度區面積明顯減少,正常細胞數量較模型2組增多;高倍鏡下染色質的邊集,核固縮較模型2組少,壞死細胞、空泡狀細胞也明顯較模型2組減少。
2.3 三組腦組織上清液中Ca2+-ATPase、LDH、CaN活性以及Ca2+、CAM水平比較
與假手術組比較,模型2組的Ca2+-ATPase活性降低,LDH、CAN活性增高,Ca2+和CAM水平增加(P均<0.05);與模型2組比較,模型1組的Ca2+-ATPase活性增高,而LDH、CaN活性降低,Ca2+和CAM水平降低(P均<0.05)。見表1。
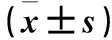
表1 三組腦組織上清液中Ca2+-ATPase、LDH、CaN活性以及Ca2+、CAM水平比較
注:與假手術組比較,*P<0.05;與模型2組比較,#P<0.05。
3 討論
腦缺血再灌注損傷具有復雜的病理生理過程,其發病機制涉及能量代謝紊亂、興奮性氨基酸毒性、細胞內Ca2+超載、自由基損傷、炎癥反應等多個環節。腦缺血再灌注時腦細胞膜受到多種因子的影響,引起一系列的分子生物學反應,使胞核濃縮,核碎裂,核溶解,細胞發生變性、壞死等表現,而且腦內細胞成片的壞死和凋亡,直接影響到神經功能表現。體外研究[2]表明,細胞內的K+對細胞凋亡具有重要作用,提高細胞培養液中的K+濃度能夠減輕甚至逆轉細胞凋亡。本研究通過對動物MCAO模型恢復再灌注的同時給予KCl溶液,觀察提高血清K+濃度能否對腦組織發揮保護作用,探討K+對大鼠腦組織缺血再灌注損傷的影響。結果顯示,模型1組的神經功能評分低于模型2組,HE染色凋亡細胞較模型2組減少。表明缺血再灌注時提高血K+濃度對腦細胞有一定的保護作用。
Ca2+是神經細胞信息傳遞的重要的第二信使,參與細胞內外的各種生物電活動和信息傳遞,維持生物的代謝功能[5]。細胞內Ca2+超載是腦缺血再灌注損傷機制的共同通路,引起神經元死亡的最后途徑[6]。Ca2+-ATPase是細胞內調節Ca2+穩態的重要酶之一,可以把Ca2+轉移至胞質以外,使得胞質內Ca2+濃度維持在較低水平[7]。當Ca2+-ATPase的活性下降時,不能及時將胞質內多余的Ca2+排出細胞外,而胞質高濃度Ca2+狀態還會使得細胞內的Ca2+庫釋放Ca2+,從而引發Ca2+超載,最終誘發一系列的級聯反應導致細胞凋亡[8~10]。Ranasinghe等[11]研究顯示,使用含K+溶液可提高Ca2+-ATPase mRNA的表達,并且提高Ca2+-ATPase mRNA的表達可以改變與心肌收縮和舒張相關的蛋白質,改善心肌收縮壓和舒張壓,從而對機體產生有益的血流動力學影響。本研究結果顯示,腦缺血再灌注損傷后,腦組織中Ca2+-ATPase活性下降,細胞內Ca2+增加,提示細胞內發生了Ca2+超載;而使用KCl調整血K+濃度后,細胞內的Ca2+-ATPase活性上升,胞質內Ca2+濃度降低。這表明提高再灌注液的血K+濃度可減輕大鼠腦缺血再灌注損傷,其機制可能與拮抗Ca2+超載有關。
CAM是目前發現的生物體內最重要的Ca2+蛋白受體,是Ca2+的細胞內靶點,CAM的激活需要Ca2+的結合。CAM與Ca2+結合后,通過調節與激酶、磷酸酶等靶蛋白的相互作用,成為Ca2+信號轉導通路的一部分[12]。而CaN是一種受Ca/CAM調節的蛋白酶,與腦內多種神經遞質有關,例如多巴胺、谷氨酸等[13]。有研究[14~16]表明,CaN升高與阿爾茨海默病、精神分裂等腦功能損害的疾病有關。本研究結果顯示,大鼠大腦發生缺血再灌注損傷后,CAM和CaN分別伴隨著胞質內Ca2+濃度升高而升高,提示出現了與Ca2+超載特異性相關的損傷。而灌注時接受KCl干預的大鼠,在24 h后CAM、CaN均明顯下降,表現出調整血K+拮抗Ca2+超載的腦保護作用。另一方面,LDH在腦細胞中廣泛存在,可以調節機體的自動代償性保護機制。腦缺血再灌注損傷發生時,腦組織損傷,釋放LDH,使腦細胞內的LDH活性增高。本研究結果顯示,模型1組的24 h LDH水平降低,表明腦損傷程度減輕。
綜上所述,腦組織缺血再灌注損傷后升高血K+濃度,能夠一定程度地減輕腦缺血再灌注損傷,為臨床采用調整血K+水平治療腦損傷提供了實驗依據。
參考文獻:
[1] Ann Panetta J, Clemens JA. Novel antioxidant therapy for cerebral ischemia-reperfusion injury[J]. Ann N Y Acad Sci, 1994,723(1):239-245.
[2] Yu SP, Yeh CH, Sensi SL, et al. Mediation of neuronal apoptosis by enhancement of outward potassium current[J]. Science, 1997,278(5335):114-117.
[3] Jia Z, Chen Q, Qin H. Ischemia-induced apoptosis of intestinal epithelial cells correlates with altered integrin distribution and disassembly of F-actin triggered by calcium overload[J]. J Biomed Biotechnol, 2012,2012:539-617.
[4] Longa EZ, Weinstein PR, Carlson S, et al. Reversible middle cerebral artery occlusion without craniectomy in rats[J]. Stroke, 1989,20(1):84-91.
[5] Bagur R, Hajnoczky G. Intracellular Ca2+sensing: its role in calcium homeostasis and signaling[J]. Mol Cell, 2017,66(6):780-788.
[6] Vannucci RC, Brucklacher RM, Vannucci SJ. Intracellular calcium accumulation during the evolution of hypoxic-ischemic brain damage in the immature rat[J]. Brain Res Dev Brain Res, 2001,126(1):117-120.
[7] Jensen TP, Buckby LE, Empson RM. Expression of plasma membrane Ca2+ATPase family members and associated synaptic proteins in acute and cultured organotypic hippocampal slices from rat[J]. Brain Res Dev Brain Res, 2004,152(2):129-136.
[8] Abas F, Alkan T, Goren B, et al. Neuroprotective effects of postconditioning on lipid peroxidation and apoptosis after focal cerebral ischemia/reperfusion injury in rats[J]. Turk Neurosurg, 2010,20(1):1-8.
[9] Warach S, Latour LL. Evidence of reperfusion injury, exacerbated by thrombolytic therapy, in human focal brain ischemia using a novel imaging marker of early blood-brain barrier disruption[J]. Stroke, 2004,35(11 Suppl 1):2659-2661.
[10] Sehgal P, Szalai P, Olesen C, et al. Inhibition of the sarco/endoplasmic reticulum (ER) Ca2+-ATPase by thapsigargin analogs induces cell death via ER Ca2+depletion and the unfolded protein response[J]. J Biol Chem, 2017,292(48):19656-19673.
[11] Ranasinghe AM, McCabe CJ, Quinn DW, et al. How does glucose insulin potassium improve hemodynamic performance? Evidence for altered expression of beta-adrenoreceptor and calcium handling genes[J]. Circulation, 2006,114(Suppl 1):1239-1244.
[12] Chin D, Means AR. Calmodulin: a prototypical calcium sensor[J]. Trends Cell Biol, 2000,10(8):322-328.
[13] Bannai H, Levi S, Schweizer C, et al. Activity-dependent tuning of inhibitory neurotransmission based on GABAAR diffusion dynamics[J]. Neuron, 2009,62(5):670-682.
[14] Kook SY, Seok Hong H, Moon M, et al. Disruption of blood-brain barrier in Alzheimer disease pathogenesis[J]. Tissue Barriers, 2013,1(2):e23993.
[15] Miyakawa T, Leiter LM, Gerber DJ, et al. Conditional calcineurin knockout mice exhibit multiple abnormal behaviors related to schizophrenia[J]. Proc Natl Acad Sci U S A, 2003,100(15):8987-8992.
[16] Ermak G, Davies KJ. Chronic high levels of the RCAN1-1 protein may promote neurodegeneration and Alzheimer disease[J]. Free Radic Biol Med, 2013,62:47-51.
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
網絡安全與數據管理(2022年1期)2022-08-29 03:15:20
導航定位學報(2022年4期)2022-08-15 08:27:00
中學生數理化·中考版(2022年8期)2022-06-14 06:55:24
新世紀智能(數學備考)(2021年9期)2021-11-24 01:14:36
成都醫學院學報(2021年2期)2021-07-19 08:35:14
新世紀智能(數學備考)(2020年9期)2021-01-04 00:25:14
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
數學物理學報(2020年2期)2020-06-02 11:29:24
光學精密工程(2016年6期)2016-11-07 09:07:19
