以精神癥狀初發的甲基丙二酸尿癥伴同型半胱氨酸血癥1例
2018-03-20 09:43:22胡文濤劉艷茹
中國實用神經疾病雜志 2018年1期
關鍵詞:癥狀
王 賞 胡文濤 劉艷茹 田 田 盧 宏
鄭州大學第一附屬醫院神經內科,河南 鄭州 450052
1 病例資料
患兒,女,12歲。2個月前家屬發現其不與同學一起玩、被動、懶散、少語,有時自言自語,說話不著邊際,有幻覺,幻視為主,似曾相識感,偶聽到有人議論自己,為非連貫性;有時感頭暈、心煩,未在意。4 d前患者出現發熱,最高達39.0 ℃,夜間突然出現口吐白沫、雙上肢抽搐,頸部僵直,持續約10 min,伴雜念多,無雙眼上翻及意識障礙,就診于我院,以“精神障礙查因:精神分裂癥?”收入精神醫學科。自發病以來,食欲、睡眠欠佳,大小便正常,體質量無增減。既往史:11 a前因誤服干燥劑行全麻手術,眼部灼傷;4 a前曾有遺尿。個人史:較同齡人反應遲鈍、記憶力、智力差。完善:CRP 5.089 mg/L;尿RT示:OB+;HB 108.0 g/L;HDL-C 0.89 mmol/L(>0.91 mmol/L);Hcy 173.62 μmol/L(4~20 μmol/L);葉酸、VitB12、肝腎功能、電解質、血糖、甲功、性激素、胸片、腹部彩超未見異常。頭顱MRI示:左側額頂葉點片狀長T1長T2信號影,FLAIR呈稍高信號,DWI未見彌散受限,考慮:脫髓鞘? 腦電圖:EEG及BEAM均輕度彌散性異常。考慮可能存在腦部器質性病變轉入我科,后精神異常加劇,主要表現為:陣發性大笑、胡言亂語、游走、發脾氣、隨意罵人、吵鬧,關系及被害妄想,自覺雙下肢以膝關節為主的疼痛;并出現雙下肢肌張力高,痙攣性截癱步態,獨立行走不能。檢查:神志清楚,精神、注意力欠佳,記憶力及計算力較差,腦神經檢查無異常,雙上肢肌張力正常,肌力4級;雙下肢肌張力高,伸肌為著,肌力遠端4級、近端3級,四肢腱反射活躍,雙側病理征自發陽性。完善:血氣分析、銅藍蛋白正常;EBV IgG (+);CMV IgG (+);肺炎支原體滴度1:320。腰穿:顱壓、常規生化、細胞學、墨汁、抗酸染色、囊蟲抗體、結核、病毒、腫瘤標志物、副腫瘤標志物抗體、堿性髓鞘蛋白、特殊細菌涂片、NMDA抗體等均未見異常;CSF電泳示白蛋白稍低,定量103.3 mg/L。復查腦電圖無異常。診療經過:疑診為病毒性腦炎,給予抗病毒治療數天。精神異常癥狀給予富馬酸喹硫平、奧氮平、丙戊酸鎂緩釋片等治療,患者精神癥狀逐漸控制。補充葉酸5 mg,tid;甲鈷胺0.5 mg,tid ;1周后復查Hcy,降至136.89 μmol/L。送檢血串聯質譜分析(MS)及尿氣相色譜質譜(GC-MS)分析(敏路思 北京 醫學檢驗所),結果:尿MMA升高104.5倍、甲基枸櫞酸水平增高1.879倍;血Ser增高,CO降低,C3水平正常。見表1。結合血清Hcy,診斷為甲基丙二酸尿癥伴同型半胱氨酸血癥。調整方案給予甲鈷胺針1 mg,qd,im;左卡尼丁1.0 g,qd,iv;甜菜堿1 g,qd;葉酸、VitB6po,乙哌立松25 mg,tid,po。治療2個月,患者雙下肢肌張力較前明顯改善,病理征仍陽性,但較前稍減弱,血Hcy明顯下降,肝腎功能無異常。
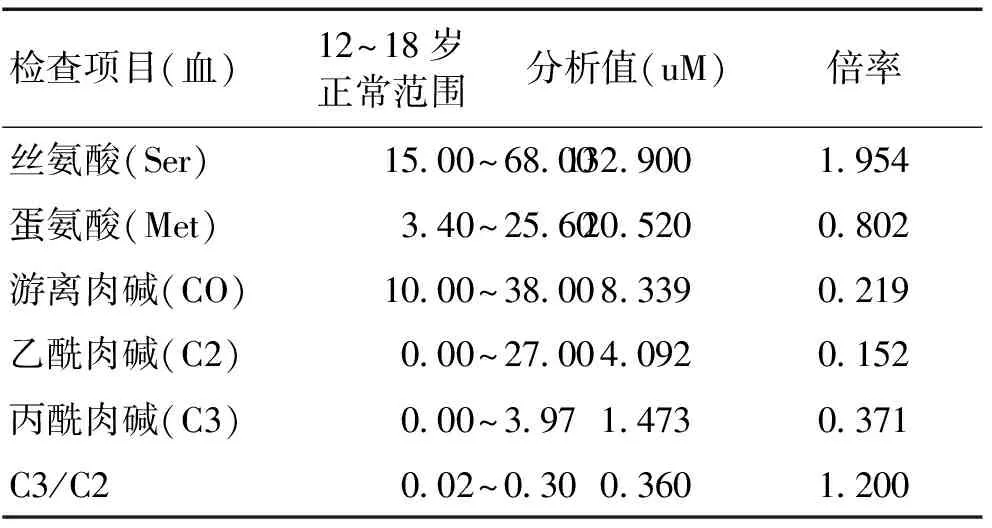
表1 血串聯質譜分析結果
2 討論
MMA因發病機制不同[1],分型較多;起病年齡各異,臨床表現多樣,具體類型需要進行基因診斷[2],多數情況下因條件有限未能進行。其臨床診斷方法[3]主要依靠臨床表現、血串聯質譜、尿氣象色譜-串聯質譜(GC-MS)檢測,合并Hcy血癥者,血清和尿液Hcy濃度增高(排除VitB12、葉酸缺乏)。其治療原則[4]是減少代謝毒物的生成及加快其代謝。急性期可給予糾酸補液,補充左旋肉堿治療;長期治療應根據對VitB12的反應性來決定治療方案,有效者一般不需特殊飲食,無效者給予特殊奶粉嚴格飲食,但應注意是否存在蛋氨酸的缺乏。MMA患者的預后取決于發病年齡、基因型、對VitB12的反應性,張堯等[5]對57例甲基丙二酸尿癥合并Hcy血癥患者進行分析,得出遲發型患者預后相對較好,約30%完全康復。WANG等[6]研究提示,對于遲發型患者,早期診斷及干預,患者的預后相對較好。
國內李荔[7]首先在2010年報道了1例精神癥狀首發的MMA患者。2012年劉曉華[8]報道了1例MMA伴Hcy血癥的雙相障礙患者,并對國外曾報道過的14例患者匯總分析發現,混合型MMA患者出現精神癥狀時易被誤診,明確診斷耗時可達數年。隨后LIU等[9]、夏德雨等[10]報道,可見臨床醫生對精神癥狀首發的MMA患者認識度明顯提高。
結合本文病例,患者有智力發育稍落后、遺尿等個人史,有精神癥狀、癲癇發作史、步態障礙等癥狀,輔助檢查提示貧血、血Hcy增高、頭顱MRI脫髓鞘改變、腦電圖異常改變,以及血串聯質譜、尿氣象色譜-質譜分析檢測,支持診斷。患兒自幼智力、記憶力較同齡人稍差,提示其可能在胎兒發育期已受到損傷[11],除有毒代謝物的神經毒性作用,甲鈷胺合成減少或缺乏導致Hcy轉變為蛋氨酸障礙,再轉變為s-腺苷蛋氨酸減少,從而導致向胎兒器官發育期的DNA和組氨酸甲基化提供的甲基基團減少。遺尿癥狀與自主神經受損相關[12]。患者的高密度脂蛋白降低,可能與MCM及其前體丙酰輔酶A蓄積,導致脂肪酸代謝異常相關。患者的精神癥狀被忽視,后因患者出現癲癇發作癥狀(未曾使用抗精神病類藥物)才引起重視,提醒臨床醫師即使在患者具有明顯的精神癥狀時,也應重視和早期識別可能存在神經系統癥狀,如本病例早期已提示可疑的智能損害,但因患者家屬未重視,未進行充分的檢查。對于本文患者的治療,因其合并Hcy血癥,試驗性治療后得知為VitB12反應型,因此給予補充甲鈷胺、左旋肉堿、甜菜堿、葉酸及Vit B6治療后癥狀得以改善,其良好的治療效果,進一步驗證了遲發型患者病程為一個相對良性過程,預后較好。因此提醒臨床工作者在今后的工作中再次遇到不明原因突發精神癥狀的青少年患者,當精神科疾病難以解釋時,需考慮到遺傳代謝性疾病的存在,早期診治。
[1] KEYFI F,TALEBI S,VARASTEH A R.Methylmalonic Acidemia Diagnosis by Laboratory Methods[J].Rep Biochem Mol Biol,2016,5(1):1-14.
[2] HUEMER M,DIODATO D,SCHWAHN B,et al.Guidelines for diagnosis and management of the cobalamin-related remethylation disorders cblC,cblD,cblE,cblF,cblG,cblJ and MTHFR deficiency[J].J Inherited metab Dis,2017,40(1):21-48.
[3] BAUMGARTNER M R,H?RSTER F,DIONISI-VICI C,et al.Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia[J].Orphanet Rare Dis,2014,9(1):130.
[4] 黃倬,韓連書,葉軍,等.甲基丙二酸血癥合并同型半胱氨酸尿癥患者治療效果分析[J].中華兒科雜志,2013,51(3):194-198.
[5] 張堯,宋金青,劉平,等.甲基丙二酸尿癥合并同型半胱氨酸血癥57例臨床分析[J].中華兒科雜志,2007,45(7):513-517.
[6] WANG X,SUN W,YANG Y,et al.A clinical and gene analysis of late-onset combined methylmalonic aciduria and homocystinuria,cblC type,in China[J].J Neurol Sci,2012,318(1-2):155-159.
[7] 李荔.甲基丙二酸血(尿)癥伴精神障礙l例[J].中國神經精神疾病雜志,2010,36(12):718-722.
[8] 劉曉華,傅燚,彭代輝.甲基丙二酸血癥伴同型半胱氨酸血癥及雙相障礙1例并文獻復習[J].中國臨床神經科,2012,20(1):78-84.
[9] LIU Y R,JI Y F,WANG Y L,et al.Clinical analysis of late-onset methylmalonic acidaemia and homocystinuria,cblC type with a neuropsychiatric presentation[J].J Neurol Neurosurg Psychiatry,2015,86(4):472-475.
[10] 夏德雨,盧碩,董秦雯,等.以精神癥狀為首發癥狀的成人型甲基戊二酸尿癥2例臨床分析及文獻復習[J].中國實用神經疾病雜志,2015,18(9):23-24.
[11] 雷如意,劉艷茹,籍煬飛,等.晚發型甲基丙二酸尿癥cblC型三例臨床特點和基因分析[J].中華神經科雜志,2014,47(2):101-106.
[12] STANGER O,FOWLER B,PIERTZIK K,et al.Homocysteine,folate and vitamin B12 in neuropsychiatrie diseases:review and treatment recommendations[J].Expert Rev Neurother,2009,9(9):1 393-1 412.
(收稿2017-05-13 修回2017-11-25)
猜你喜歡
初中生學習指導·提升版(2023年8期)2023-09-12 10:26:19
保健醫苑(2022年1期)2022-08-30 08:39:40
中老年保健(2021年12期)2021-08-24 03:30:44
今日農業(2020年17期)2020-10-27 03:10:52
今日農業(2020年16期)2020-09-25 03:05:08
家庭醫學(下半月)(2020年2期)2020-05-11 02:07:18
基層中醫藥(2020年10期)2020-02-13 15:45:52
吉林蔬菜(2017年10期)2017-11-01 07:47:04
獸醫導刊(2016年6期)2016-05-17 03:50:35
中國醫學影像學雜志(2015年9期)2015-12-15 11:03:26
