二代測序檢測乳腺癌易感基因在乳腺癌發病風險預測及臨床治療中的應用
2017-12-11 11:39:42吳楠于津浦趙晶趙洋穆坤張軍金釗劉俊田
中國腫瘤臨床 2017年20期
吳楠 于津浦 趙晶 趙洋 穆坤 張軍 金釗 劉俊田
二代測序檢測乳腺癌易感基因在乳腺癌發病風險預測及臨床治療中的應用
吳楠①于津浦②趙晶①趙洋①穆坤①張軍①金釗①劉俊田①
目的:通過二代測序技術篩選乳腺癌易感基因突變位點,探討其對乳腺癌風險預測、臨床干預及預后指導的意義。方法:采取2013年11月至2015年7月272例就診天津醫科大學腫瘤醫院的乳腺癌患者146例、高危人群71例及健康者55例3組樣本的外周血,采用二代測序技術進行DNA檢測。采用Amplicon方法篩選BRCA1、BRCA2、PTEN、STK11、TP53及BAP1全外顯子區域有價值的突變位點,分析3組的基因突變發生率及乳腺癌患者易感基因突變與臨床病理特征之間的相關性。結果:經篩選獲得177個突變位點,去重后共得到67個突變位點,其中包括50個單核苷酸突變(single nucleotide variants,SNVs)、8個無義突變(nonsense mutation)和9個插入缺失突變(insertion-deletions,InDels)。3組突變位點中31個突變位點收錄于ExAC數據庫,40個突變位點收錄于Clin Var數據庫。本研究中21個新發現的突變位點在ExAC、Clin Var或db SNP數據庫中均未提及。統計分析發現85.1%(57/67)的突變發生于乳腺癌患者及高危人群中,且突變陽性乳腺癌患者有較高的淋巴結轉移率(P=0.010)及病理分期(P=0.002),致病性突變的乳腺癌患者的腫瘤家族史(P=0.005)及三陰性乳腺癌比例(P=0.009)均高于非致病性突變者。結論:乳腺癌易感基因突變位點在乳腺癌風險預測、臨床治療及預后評價方面具有重要意義。
二代測序 乳腺癌易感基因 風險預測 臨床干預 預后評價
乳腺癌早期預防是其發病率降低的主要原因[1]。流行病學調查顯示,乳腺癌易感基因突變主要表現在其家族的聚集性[2],其中二代測序技術對檢測乳腺癌易感基因突變具有重要意義。本研究旨在對BRCA1、BRCA2、PTEN、TP53、STK11和BAP1乳腺癌易感基因進行測序,分析其在乳腺癌風險預測、臨床治療及預后評價中的意義。
1 材料與方法
1.1 材料
1.1.1 病例資料 采取2013年11月至2015年7月272例于天津醫科大學腫瘤醫院診治的146例乳腺癌患者、71例高危人群及55例健康者的外周血。入選標準:1)患者:同意入組的乳腺癌患者;2)高危人群:既往有腫瘤病史(已排除皮膚基底細胞癌、宮頸原位癌等早期癌)或家族中2位及以上一級親屬患惡性腫瘤(年齡≤50歲);3)健康人群:既往無腫瘤病史及腫瘤家族史。本研究得到天津醫科大學腫瘤醫院倫理委員會批準,且患者均簽署知情同意書。
1.1.2 試劑 Control DNA ACD1、Control Oligo Pool ACP1、CAT、OHS1、SW1、UB1、TDP1/PMM2 PCR擴增預混液、LNA1/LNB1混合試劑、NW1均購自美國Illumina公司。
1.2 方法
1.2.1 標本采集 采取患者外周靜脈血5mL,1200r/min離心10 min后,取中間懸浮層白膜200 μL。
1.2.2 DNA提取 吸取20 μL蛋白酶K、200 μL全血樣本及200 μL緩沖液AL至1.5 mL離心管混勻,56℃孵育10min,短暫離心后加入200μL無水乙醇混勻,8000r/min離心1 min,棄濾液。分別加入500 μL緩沖液AW1、AW2混勻,8000r/min及14000r/min短暫離心后,加入200μL緩沖液AE,室溫孵育1 min,8 000 r/min離心1 min。
1.2.3 DNA濃度及純度測量 初始化儀器后,取1.5 μL去離子水作為空白對照,依次加入1.2 μL的DNA提取樣本檢測其OD值,保留OD260/280為1.8~2.0、濃度為50 ng/μL及以上的樣本。
1.2.4 DNA文庫制備 實驗方法參照美國Illumina公司的TruSeq Amplicon Cancer Panel說明書操作[3]。
1.2.5 突變位點致病性檢測 通過SIFT、Polyphen2、LRT、Mutation Taster、Mutation Assessor、FATHMM、Meta SVM和Meta LR軟件對篩選出的每一個突變位點進行檢測。當有1個軟件預測某個突變位點為功能缺陷時記為1分,通過ROC曲線得出能夠預測致病性突變的cut off值為2.5分。本研究將3組樣本中突變位點評分≥3分時定為致病性突變,對于插入缺失突變(insertion-deletions,InDels)和無義突變(nonsense mutation)采用Mutation Taster進行評價。
1.3 統計學分析
采用SPSS 20.0軟件進行統計學分析。計量資料采用t檢驗;計數資料采用χ2檢驗。P<0.05為差異具有統計學意義。
2 結果
2.1 樣本測序數據的質量統計
3組樣本測序深度為200~1 000×(圖1A);92%樣本的Q30超過80%(圖1B);讀取至目標區域的數據超過85%(圖1C);編碼區的覆蓋率達到95%以上(圖1D);樣本間測序深度保持一致(圖1E);不同基因所占數據量比例穩定(圖1F)。
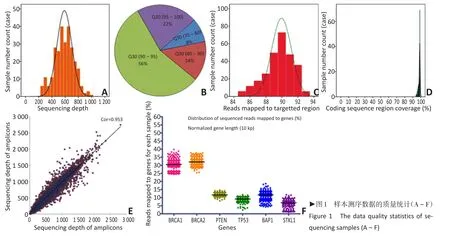
圖1 樣本測序數據的質量統計(A~F)Figure 1 The data quality statistics of sequencing samples(A~F)
2.2 樣本突變位點的篩選流程
通過圖2所示篩選的方式共獲得177個突變位點,其中包含158個單核苷酸突變(single nucleotide variants,SNVs)、8個無義突變和11個InDels。去重后為67個突變位點,包括50個SNVs、8個無義突變和9個InDels。突變位點與ExAC、ClinVar數據庫收錄相同的為31、40個,本研究新發現的21個突變位點在ExAC,Clin Var或dbSNP數據庫中均未提及。
2.3 乳腺癌易感基因突變分布及風險預測
本研究最終發現的67個基因突變中50個(74.6%)發生于BRCA1/2,46個分別收錄于ExAC,ClinVar或dbSNP數據庫。46個突變中22個(47.8%)為致病性突變,其中5個為InDels、6個為無義突變、11個為錯義突變。本研究新發現的21個突變位點中14個(66.7%)為致病性突變,其中3個為InDels、2個為無義突變、9個為錯義突變。
2.4 檢測樣本中乳腺癌易感基因的分布情況
本研究行外周血檢測的272例患者中攜帶突變的為164例,其中乳腺癌患者70例、高危人群49例、健康者45例。乳腺癌患者的突變位點為42個,高危人群為22個,健康者為10個,高達85.1%(57/67)突變發生于乳腺癌患者及高危人群。146例乳腺癌患者中9例臨床病理資料缺失,137例乳腺癌患者中易感基因突變及易感基因致病性突變與臨床病理特征的關系見表1。
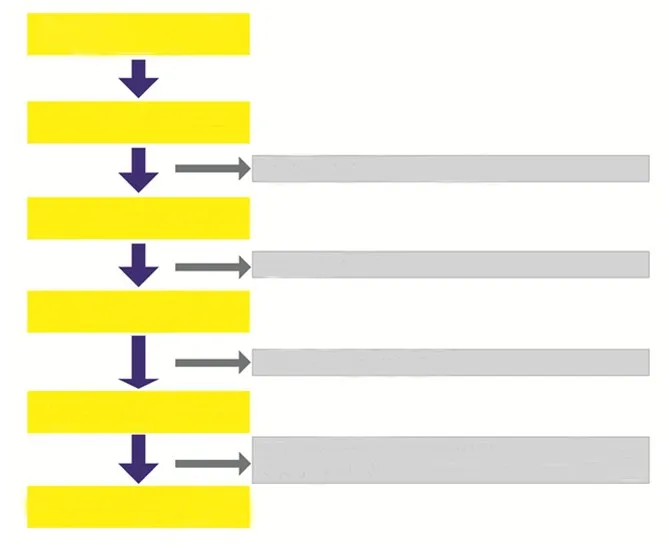
圖2 突變位點的篩選流程Figure 2 Flow chart of screening 177 variants
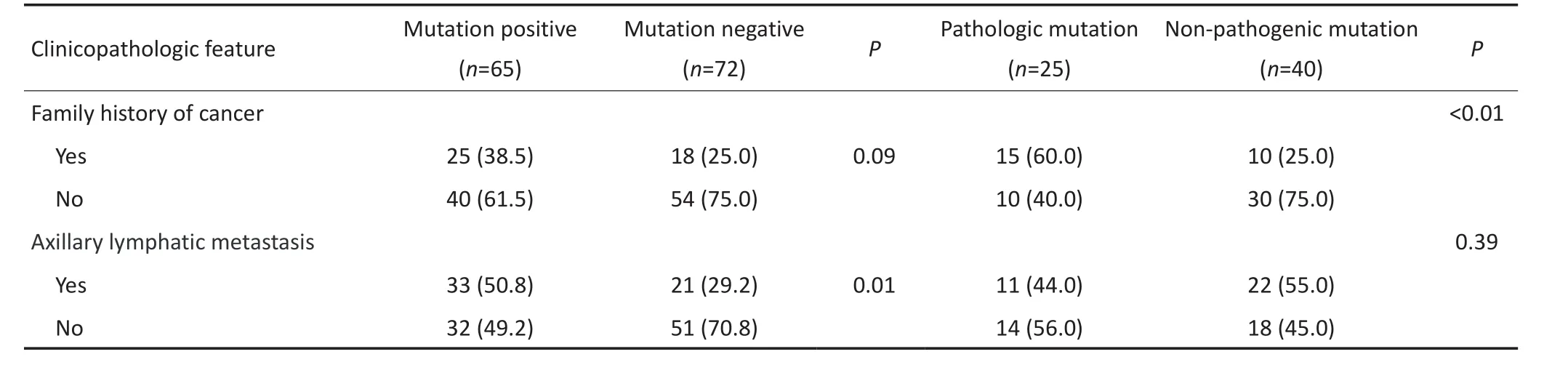
表1 6個乳腺癌易感基因突變及易感基因致病性突變與乳腺癌患者臨床病理特征的關系Table 1 Relationship of breast cancer susceptibility gene mutation and pathologic mutation with clinicopathologic feature of patients
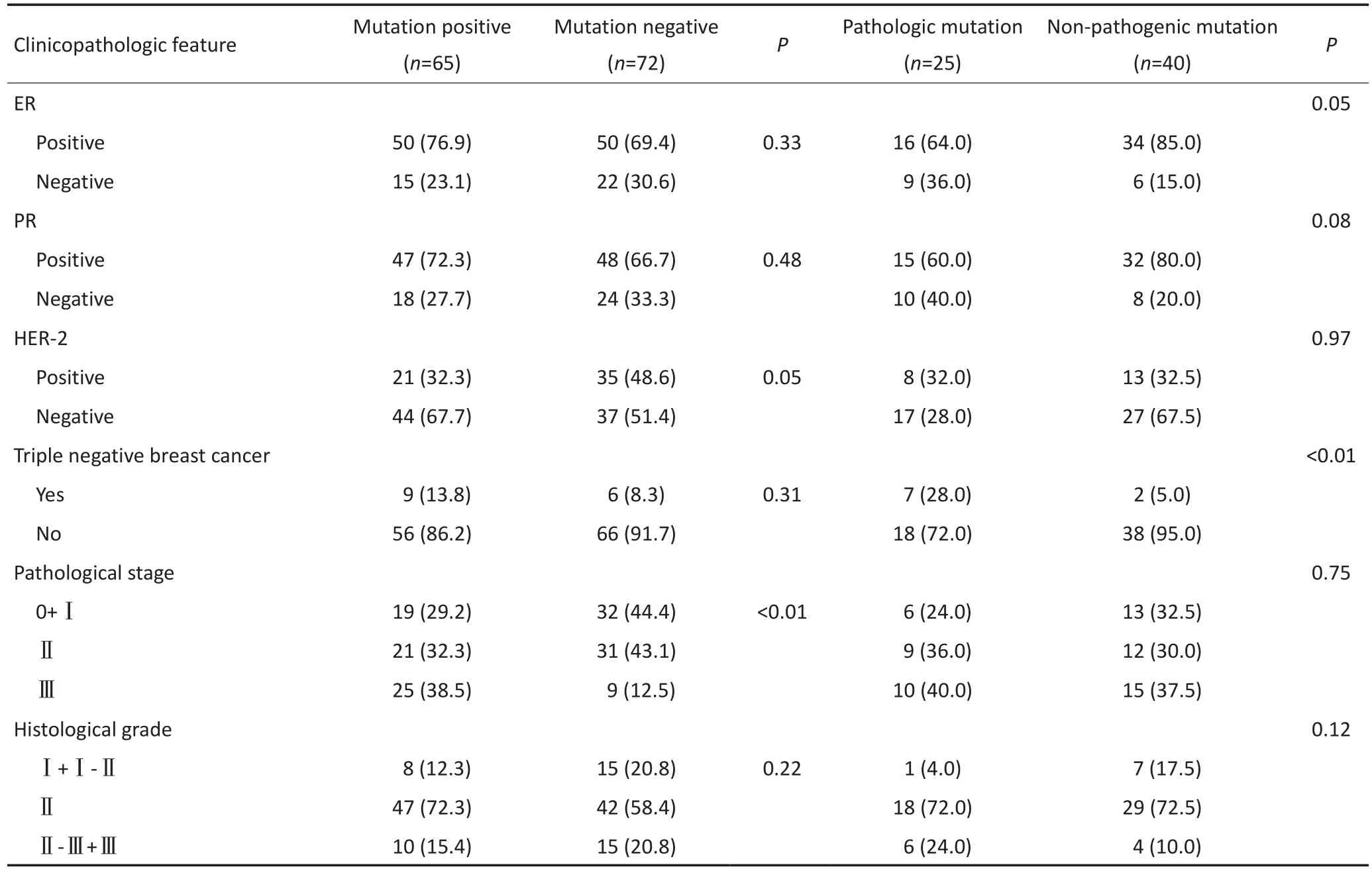
表1 6個乳腺癌易感基因突變及易感基因致病性突變與乳腺癌患者臨床病理特征的關系(續表1)Table 1 Relationship of breast cancer susceptibility gene mutation and pathologic mutation with clinicopathologic feature of patients
3 討論
研究表明乳腺癌易感基因突變是乳腺癌具有家族聚集性的重要原因[4],其中BRCA1/2是較為重要的兩個基因,攜帶BRCA1/2突變的70歲前健康女性罹患乳腺癌的累計風險高達55%~85%[5],TP53、PTEN、STK11、CDH1、ATM、CHEK2乳腺癌易感基因也與乳腺癌發生發展密切相關[6]。
本研究通過二代測序技術篩選乳腺癌易感基因全外顯子區域突變位點,與傳統的一代測序相比二代測序的優勢是更加高效[7]。目前,乳腺癌易感基因檢測因缺乏熱點區,所檢測的基因片段較長,選擇傳統的檢測技術效果不佳,因此二代測序技術備受矚目。同時,二代測序技術也存在不足之處,如檢測步驟較繁瑣,數據分析難度大,并且存在大量未知位點的判讀等。基于以上優缺點,二代測序技術仍得到國內外學者的廣泛應用[8-9]。本研究測序結果中得出測序數據質量高、目標區域測序深度均勻、目標區域覆蓋度好、擴增效率穩定、樣本間測序深度重復性高、樣本間堿基變化分布均勻、突變圖譜符合Germline Mutation特征的優勢。
本研究通過篩選得到67個突變位點,其中46個突變(68.7%)收錄于ExAC、ClinVar或dbSNP數據庫,由此證實本研究結果的可信度,21個突變位點(31.3%)為本研究中新發現的突變位點,且均發生于乳腺癌患者或高危人群,由此說明這些新發現的突變位點可作為預測乳腺癌發病風險的新切入點,為進一步的研究提供理論依據。統計分析發現,乳腺癌易感基因突變在乳腺癌患者及高危人群中的發生率為85.1%(57/67),明顯高于健康者的1.5%(1/67),提示檢測乳腺癌易感基因突變位點可為預測乳腺癌發生風險、提高早期乳腺癌的檢出率提供幫助。但需要指出的是,篩選出的突變位點需要更多的研究來驗證,從而建立安全有效的乳腺癌危險預測模型,達到指導乳腺癌危險評估的目的。
本研究進一步分析發現,突變陽性患者的腋下淋巴結轉移比例較高(P=0.010),且病理分期中Ⅲ期患者所占比例較大(P=0.002),由此提示發生易感基因突變的乳腺癌患者預后可能更差,對攜帶乳腺癌易感基因突變患者需要考慮是否適當強化臨床治療。研究表明,BRCA1突變的乳腺癌中高達70%患者為三陰性乳腺癌,BRCA2突變的乳腺癌中16%~23%患者為三陰性乳腺癌[10],本研究發現在6個乳腺癌易感基因突變的患者中致病性突變患者的三陰性乳腺癌所占比例高(P=0.009),與上述研究結果一致。
綜上所述,本研究通過二代測序技術檢測乳腺癌易感基因突變情況,從而為乳腺癌早期預防,臨床治療及預后評價提供一定理論依據,但由于本研究樣本例數較少,且未進行相關生存分析,因此其對于乳腺癌患者治療及預后的指導意義有待進一步證實。
[1]Hooks MA.Breast cancer:risk assessment and prevention[J].South Med J,2010,103(4):333-338.
[2]Collaborative Group on Hormonal Factors in Breast Cancer.Familial breast cancer:collaborative reanalysis of individual data from 52 epidemiological studies including 58,209 women with breast cancer and 101,986 women without the disease[J].Lancet,2001,358(9291):1389-1399.
[3]Simen BB,Yin L,Goswami CP,et al.Validation of a next-generationsequencing cancer panel for use in the clinical laboratory[J].Arch Pathol Lab Med,2015,139(4):508-517.
[4]Hedenfalk IA,Ringnér M,Trent JM,et al.Gene expression in inherited breast cancer[J].Adv Cancer Res,2002,84:1-34.
[5]Hall MJ,Reid JE,Burbidge LA,et al.BRCA1 and BRCA2 mutations in women of different ethnicities undergoing testing for hereditary breast-ovarian cancer[J].Cancer,2009,115(10):2222-2233.
[6]Zhang B,Beeghly-Fadiel A,Long J,et al.Genetic variants associated with breast-cancer risk:comprehensive research synopsis,metaanalysis,and epidemiological evidence[J].Lancet Oncol,2011,12(5):477-488.
[7]Hong H,Zhang W,Shen J,et al.Critical role of bioinformatics in translating huge amounts of next-generation sequencing data into personalized medicine[J].Sci China Life Sci,2013,56(2):110-118.
[8]Jalkh N,Chouery E,Haidar Z,et al.Next-generation sequencing in familial breast cancer patients from Lebanon[J].BMC Med Genomics,2017,10(1):8.
[9]Kluska A,Balabas A,Paziewska A,et al.New recurrent BRCA1/2 mutations in Polish patients with familial breast/ovarian cancer detected by next generation sequencing[J].BMC Med Genomics,2015,8:19.
[10]Stevens KN,Vachon CM,Couch FJ.Genetic susceptibility to triplenegative breast cancer[J].Cancer Res,2013,73(7):2025-2030.
(2017-02-03收稿)
(2017-09-28修回)
Second generation sequencing detection breast cancer susceptibility gene variants for risk prediction and clinical treatment
Nan WU1,Jinpu YU2,Jing ZHAO1,Yang ZHAO1,Kun MU1,Jun ZHANG1,Zhao JIN1,Juntian LIU1
1Department of Breast Cancer,2Department of Biocherapy,Tianjin Medical University Cancer Institute and Hospital;National Clinical Research Center for Cancer,Key Laboratory of Cancer Prevention and Therapy,Tianjin;Tianjin's Clinical Research Center for Cancer,Tianjin 300060,China;Tianjin Medical University Cancer Institute and Hospital;National Clinical Research Center for Cancer,Key Laboratory of Cancer Prevention and Therapy,Tianjin;Tianjin's Clinical Research Center for Cancer,Tianjin 300060,China
Juntian LIU;E-mail:ljt641024@163.com
Objective:To investigate the function of breast cancer susceptibility gene variants in predicting breast cancer risk and guiding clinical treatment through DNA sequencing.Methods:This study involved 146 patients,71 high-risk cases,and 55 healthy people,totaling 272 cases.The subjects were treated in Tianjin Medical University Cancer Institute and Hospital from November 2013 to July 2015.Genomic DNA was sequenced by a second generation sequencing platform.All exon areas of six common breast cancer susceptibility genes(BRCA1,BRCA2,PTEN,STK11,TP53,and RAP1)were sequenced through amplicon sequencing method.Meaningful variants including single nucleotide variants(SNVs),insertion-deletions(InDels)and nonsense mutations were selected and statistical methods,such as t test and χ2test,were used to analyze the statistical differences in incidence rates among three groups.Results:A total of 177 meaningful variants were confirmed,including 50 SNVs,8 nonsense mutations,and 9 InDels.Among the variants,31 were recorded in the Exome Aggregation Consortium(ExAC),40 were noted in ClinVar database,and 21 were not encoded in the present database,which were defined as new variants in this study.Conversely,57 variants(85.1%)were found in breast cancer patients and high-risk cases,and the incidence of axillary lymph node metastasis(P=0.010)and pathological stages(P=0.002)in mutation positive patients were both higher than mutation negative patients.Moreover,the percentage of family history of cancer(P=0.005)and triple negative breast cancer(P=0.009)were both higher in patients carrying pathogenic mutations than in nonpathogenic patients.Conclusion:Breast cancer susceptibility gene variants may not only be a tool used to predict the risk of getting breast cancer but also a meaningful guideline for the clinical treatment and prognosis evaluation.
:second generation sequencing,breast cancer susceptibility gene,risk prediction,clinical intervention,prognosis evaluation
10.3969/j.issn.1000-8179.2017.20.203
①天津醫科大學腫瘤醫院乳腺二科,國家腫瘤臨床醫學研究中心,天津市腫瘤防治重點實驗室,天津市惡性腫瘤臨床醫學研究中心,乳腺癌防治教育部重點實驗室(天津市300060);②生物治療科
劉俊田 ljt641024@163.com

吳楠 專業方向為乳腺腫瘤預防及臨床治療等。E-mail:1090815242@qq.com
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中老年保健(2022年6期)2022-08-19 01:41:48
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中國生殖健康(2020年2期)2021-01-18 02:51:26
中國生殖健康(2019年2期)2019-08-23 08:11:42
中國生殖健康(2019年6期)2019-01-06 09:20:12
小學生導刊(2018年13期)2018-06-29 03:49:00
祝您健康(2018年5期)2018-05-16 17:10:16
